 |
|
|
|
|
Research Projects:: Dynamics for Reactions of Ion Pairs in WaterCarbon atoms in organic molecules are most often neutral. Positively charged carbocations have attracted the interest of synthetic organic chemists, because of their use as intermediates in reactions leading to formation of carbon-carbon bonds. Our work on carbocations had focused on defining the stability of these species as intermediates of solvolysis reactions, through the determination of rate and equilibrium constants for these stepwise reactions (Scheme 1). This has led to the development of experimental methods to characterize these parameters for carbocations that are sufficiently stable to form in aqueous solution. 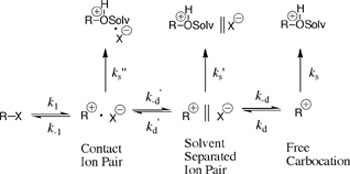
It is convenient to separate heterolytic bond cleavage and bond formation (k1 and k-1) from the transport steps (k-d', k-d, kd' and kd) for Scheme 1. The values of the rate constants for bond cleavage and formation depend strongly on substrate structure and are generally similar, respectively, to the experimental rate constants for solvolysis of R-X and capture of the carbocation reaction intermediate R+ by X-, respectively. By comparison, the rate constants for reversible separation of ion pairs to free ions show a much smaller dependence on the structure of the ions and a significant dependence on the solvent. Contact and solvent-separated ion pairs form whenever solvolysis proceeds to the free carbocation. However, these intermediates are generally only thought of as "significant" when their formation can be detected by experiment. Our work has focused on the detection and analysis of "signature" of ion pair reactions with the specific goal of determining absolute rate constants for these processes. This is a challenging problem given the ephemeral nature of ion pairs in water and their short short lifetimes of ca. 0.10 nsec. Our published work in this area includes an examination of the following reactions of ion pairs: (1) The addition of solvent to a carbocation paired with its leaving group anion (ks" and ks', Scheme 1). The effect of direct addition of solvent to an ion-pair reaction intermediate is to cause the rate of solvolysis to become faster than for a reaction where products form exclusively by addition of solvent to the unpaired carbocation (ks), and it is possible to detect this as a deviation from a rate law for the latter reaction. See Journal of Organic Chemistry, 57, 625-629 (1992). (2) Protonation of the leaving group anion, which prevents internal return of the ion pair to reactant, and has the effect of make substrate ionization irreversible. [Unpublished] (3) Racemization of a chiral ion pair, or bond rotation of the leaving group ion followed by internal return that leads to formation of a product that has undergone racemization or isomerization with exchange or reacting atoms at the leaving group. Our work on ion-pair isomerization is exemplified by the following study. 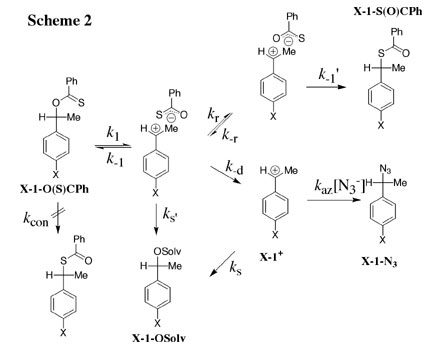
The reactions of ring-substituted 1-phenylethyl thionobenzoates X-1-O(S)CPh (X = 4-MeS, 4-F, and 4-Me) in 50:50 (v/v) trifluoroethanol/water at 25 C and I = 0.50 (NaClO4) proceed by a stepwise mechanism through ion pair intermediates X-1+*-O(S)CPh which partition between diffusional separation to free ions (k-d), direct nucleophilic addition of solvent (ks'), and ion pair reorganization (kr) followed by fast collapse to the isomerization products, 1-phenylethyl thiolbenzoates X-1-S(O)CPh. Combination of the product rate constant ratios kr/(ks' + k-d) with previously reported values of ks' and k-d gives kr = 1 x 1011/s for reorganization of the ion pairs within an aqueous solvation shell. See (a) Journal of the American Chemical Society, 122, 3963-3964 (2000), (b) Organic Letters, 3, 1237-1240 (2001) and (c) Journal of Physical Organic Chemistry, 16, 484-490 (2003). |
| |||||||||
All material © John Richard 2003-2004 | Website Developed by Iserloh Design | ||||||||||