 |
|
|
|
|
Research Projects:: Formation and Stability of Carbanions in Water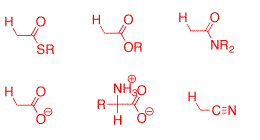
As of 1992 the kinetic and thermodynamic barriers for deprotonation of weak carbon acids in aqueous solution had not well characterized! Our goal at this time was to develop methods for the determination of the pKas in water of carbon acids stabilized by simple functional groups and to use these data to gain new insight into the mechanism for proton transfer at carbon. 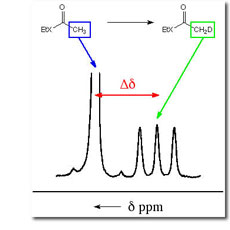
The first step was the development of an improved method using 1H NMR for monitoring carbanion formation by following exchange of hydrogen from carbon acids for deuterium from solvent by taking advantage of the 2H perturbation of 1H chemical shifts. The Figure to the right shows that the isotope exchange of ethyl thioacetate leads to disappearance of the singlet due to the α-CH3 group of unexchanged thioester acetate, and appearance of an upfield triplet due to the α-CH2D group of monodeuteriated thioester, in which the remaining α-protons are coupled to the α-deuterium (JHD = 2.2 Hz). We have used these methods to determine the pKas for a wide variety of simple carbon acids. (1) Esters. The rate constants for deprotonation of ethyl acetate by 3-substituted quinuclidines are correlated by b = 1.09 � 0.05. The limits of kBH = kBH = 2 - 5 x 109 M-1 s-1 for the encounter-limited reaction of the simple oxygen ester enolate with protonated quinuclidine (pkBH = 11.5) were combined with kB = 2.4 x 10-5 M-1 s-1 for deprotonation of ethyl acetate by quinuclidine, to give pKaK = 25.6 � 0.5 for ionization of ethyl acetate as a carbon acid in aqueous solution. A rate-equilibrium correlation for proton transfer from methyl and benzylic monocarbonyl compounds to hydroxide ion has been extended by 6 pK units in the thermodynamically unfavorable direction, and it is shown that the absence of curvature of this correlation is inconsistent with a constant Marcus intrinsic barrier for the enolization of simple carbonyl compounds. See Journal of the American Chemical Society, 118, 3129-3141 (1996). 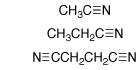
(2) Nitriles. Rate constants kDO (M-1 s-1) for the deprotonation of cyanoalkanes by deuteroxide ion in D2O at 25 C were determined by following the appearance of the deuterium-labelled cyanoalkanes by 1H NMR. These data were evaluated to give the following pKas in water: CH3CN, 28.9; CH3CH2CN, 30.9; NCCH2CH2CN, 26.6. High level ab initio calculations on cyanoalkanes and α-cyano carbanions and combined QM/Monte Carlo calculations of their free energies of solvation were carried out. The interaction between a carbanionic center and an α-cyano substituent is concluded to be largely polar. The 5.1-fold difference in α-cyano and β-cyano substituent effects on carbon acidity in water which, nominally, is consistent with significant resonance stabilization of α-cyano carbanions, is attributed to the differential solvation of cyanoalkanes and cyanocarbanions. The free energy change for the highly unfavorable tautomerization of acetonitrile to ketenimine in water was computed as DGT = 30.7 kcal/mol. We have proposed that the large instability of the ketenimine cumulative double bond favors the valence bond resonance form of the α-cyanocarbanion in which there is a formal carbon-nitrogen triple bond and the negative charge is localized at the α-carbon. See Journal of the American Chemical Society, 121, 715-726 (1999). 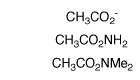
(3) Amides and carboxylate ions. Second-order rate constants were determined in D2O for deprotonation of acetamide, N,N-dimethylacetamide and acetate anion by deuteroxide ion and for deprotonation of acetamide by quinuclidine. The values of kB = 4.8 x 10-8 M-1 s-1 for deprotonation of acetamide by quinuclidine (pkBH = 11.5) and kBH = 2 - 5 x 109 M-1 s-1 for the encounter-limited protonation of the enolate by protonated quinuclidine give pKaC = 28.4 for ionization of acetamide as a carbon acid. The limiting value of kHOH = 1 x 1011 s-1 for protonation of the enolate of acetate anion by solvent water and kHO = 3.5 x 10-9 M-1 s-1 for deprotonation of acetate anion by HO- give pKa = 33.5 for acetate anion. The change in rate-limiting step from chemical proton transfer to solvent reorganization results in a downward break in the slope of the plot of log kHO against carbon acid pKa for deprotonation of a wide range of neutral α-carbonyl carbon acids by hydroxide ion, from -0.40 to -1.0 . Good estimates are reported for the stabilization of the carbonyl group relative to the enol tautomer by electron-donation from α-SEt, α-OMe, α-NH2 and α-O- substituents. The α-NH2 and α-OMe groups show similar stabilizing interactions with the carbonyl group, while the interaction of α-O- is only 3.4 kcal/mol more stabilizing than for α-OH. We propose that destabilization of the enolate intermediates of enzymatic reactions results in an increasing recruitment of metal ions by the enzyme to provide electrophilic catalysis of enolate formation. See Journal of the American Chemical Society, 124, 2957-2968, (2002). 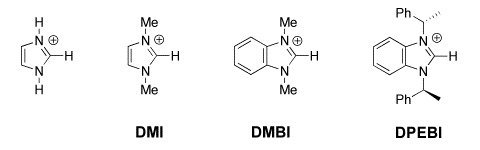
(4) Imidazolium Ions. second-order rate constants kDO (M-1 s-1) were determined for exchange for deuterium of the C(2)-proton of a series of simple imidazolium cations to give the corresponding imidazol-2-yl carbenes in D2O at 25 C and I = 1.0 (KCl). Evidence is presented that the reverse protonation of imidazol-2-yl carbenes by solvent water is limited by solvent reorganization and occurs with a rate constant of kHOH = kreorg = 1011 s-1. The data were used to calculate reliable carbon acid pKas for ionization of imidazolium cations at C(2) to give the corresponding singlet imidazol-2-yl carbenes in water: pKa = 23.8 for the imidazolium cation, pKa = 23.0 for the 1,3-dimethylimidazolium cation, pKa = 21.6 for the 1,3-dimethylbenzimidazolium cation, and pKa = 21.2 for the 1,3-bis-((S)-1-phenylethyl)benzimidazolium cation. The data also provide the thermodynamic driving force for a 1,2-hydrogen shift at a singlet carbene: K12 = 5 x 1016 for rearrangement of the parent imidazol-2-yl carbene to give neutral imidazole in water at 298 K, which corresponds to a favorable Gibbs free energy change of 23 kcal/mol. We present a simple rationale for the observed substituent effects on the thermodynamic stability of N-heterocyclic carbenes relative to a variety of neutral and cationic derivatives that emphasizes the importance of the choice of reference reaction when assessing the stability of N-heterocyclic carbenes. See Journal of the American Chemical Society, 126, 4366-4374, (2004). There are many more simple carbon acids whose pKas might be determined using these methods, including sulfonium ions, phosphonium ions, alkyl sulfones and alkyl sulfoxides. |
| |||||||||
All material © John Richard 2003-2004 | Website Developed by Iserloh Design | ||||||||||