 |
|
|
|
|
Research Projects:: Catalysis of Phosphate Diester Hydrolysis by Metal Ion ComplexesStudies on catalysis of RNA-hydrolysis by metal-ion complexes have identified many active catalysts, but have provided only limited insight into the relationship between the catalyst structure and the activity towards cleavage of phosphate diesters. Consequently, the results from this work allow qualitative comparisons of the activity for different catalysts, but do not provide the type of data needed to formulate a mechanism for the stabilization of the catalyst-bound transition state. Our aim is to develop systematic methods for quantitative analyses of the catalytic activity of these metal ion complexes, in order to rationalize why different catalytic activities are observed for structurally related catalysts. This question is of considerable intellectual interest, and its resolution will provide insight into reaction mechanisms that can be used in the design of catalysts with enhanced activity. Our work on these problems studies have produced results which provide a detailed description of the catalytic reaction mechanism along with interesting leads that will guide the design of new catalysts. 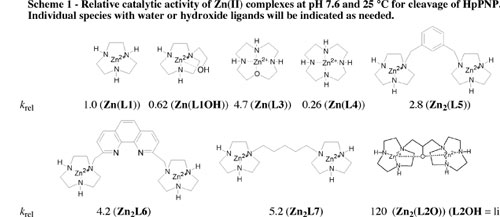
(1) We have shown that the two metal-ion centers in Zn2(L2O) function cooperatively in the cleavage of simple phosphodiesters and RNA. This conclusion follows directly from the comparison of the relative catalytic activities of a series of dinuclear catalysts of HpPNP cleavage at pH 7.6 and 25 C (Scheme 1). This Scheme shows that the total activity of several dinuclear catalysts [Zn2(L5), Zn2(L6) and Zn2(L7)] is generally not much greater than the sum of their parts [Zn(L1)]. By comparison, the dinuclear catalyst Zn2(L2O) shows a catalytic activity that is 120-times greater than observed for Zn(L1), or 60-fold greater than the activity expected for a complex in which the tethered macrocycles react independently. See Inorganic Chemistry, 42, 7737-7746 (2003). 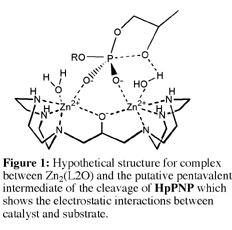
(2) We have shown that the pendant hydroxyl group of Zn(L1OH) is protonated at pH 7, but that the linker hydroxyl group at Zn2(L2O) is ionized at neutral pH. Two lines of evidence provide support for this conclusion: (A) Potentiometric titrations show that there is a pKa of < 6 for an acidic oxygen in the formation of Zn2(L2O), but not for Zn(L1OH). 1H NMR spectra show that the chemical shift of the signals for the protons closest to the relevant hydroxyl groups of Zn2(L2O) and Zn(L1OH) remain fixed as the pH is increased from = 7-10. (B) The X-ray structure of [Zn2(L2O)(Cl)(H2O)2](ClO4)2 for a crystal obtained at pH 6 features a bridging alkoxide that forms a chelate to the two Zn(II) ions. The two Zn(II) ions have different coordination numbers and geometries in spite of the symmetry of the ligand L2OH. This highlights the structural flexibility of Zn(II). By comparison, the crystal structure of [Zn(L1OH)(Br)](Br) obtained at pH 9.1 confirms that a neutral alcohol group is coordinated to Zn(II). These data show that the bridging alkoxide group of Zn2(L2O) shields the electrostatic interactions between the Zn(II) ions, and allows the cations to be drawn close together in a complex of greatly enhanced activity (Figure 1). This high density of positive charge at Zn2(L2O) is ideal for providing electrostatic stabilization of the transition state for cleavage of phosphodiesters relative to the reactant state, because there is a net unit increase in negative charge on proceeding from the reactant to transition state. See Journal of the American Chemical Society, 125, 1988-1993 (2003). 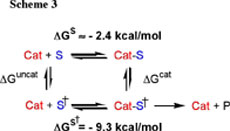
(3) We have characterized the substrate specificity of Zn2(L2O) for cleavage of nitrophenyl phosphate diesters. A comparison of the observed first-order rate constants for hydroxide ion-catalyzed cleavage, and the second-order rate constant for Zn2(L2O)-catalysed cleavage at pH of 7.0 shows that the rate acceleration from catalysis by 1 M of Zn2(L2O) is 50-fold larger for cleavage of HpPNP (9.8 x 106-fold) than for cleavage of UpPNP (1.8 x 105-fold). This corresponds to 9.3 kcal/mol stabilization of the transition state for cleavage of the minimal substrate HpPNP by interaction with Zn2(L2O) (Scheme 3, S = HpPNP) and a smaller 7.1 kcal/mol stabilization of the transition state for cleavage of the nucleoside substrate UpPNP. The observation that the transition state for cleavage of HpPNP is more strongly stabilized (tightly bound) by Zn2(L2O) than for cleavage of UpPNP is surprising and revealing because, while the opportunity for development of binding interactions to the nucleoside substrate UpPNP transition state is greater than that for the minimal substrate HpPNP, the observed interactions are significantly weaker. Intramolecular tethering of the metal ions at the macrocyclic ligands across the bridging alkoxide ion of Zn2(L2O) has the effect of generating a highly charged core of unusual catalytic activity. These results provide evidence for the notion that a significant drawback of this array is that access to the catalytic core is restricted, so that HpPNP may bind closely to the cationic core to achieve stabilization of the anionic transition state, while the interaction of UpPNP with the catalyst is not as effective, perhaps due to steric interactions between the catalyst and nonreacting portions of this substrate. See Journal of the Chemical Society, Chemical Communications, 2832-2833 (2003). (4) We have determined the effect of changing the metal cation on the activity of dinuclear complexes of L2OH towards cleavage of HpPNP. The relative reactivity of dinuclear Zn(II), Cu(II) and Cd(II) complexes of L2OH have been rationalized by a consideration of the different geometric preferences of complexes of the metal cations, and the relative Lewis acidities of their complexes. A comparison of the X-ray crystal structures Zn2(L2O) and Cu2(L2O) provides evidence that the difference in the catalytic activity of these two complexes (Figure 4) is due to the larger number of open coordination sites in the active Zn(II) complex compared to the Cu(II) complex for interaction with the substrate. The subtle differences in the catalytic properties of the dinuclear Cd(II) and Zn(II) complexes are a consequence of the higher Lewis acidity of Zn(II) bound water compared to Cd(II) bound water. See Inorganic Chemistry 43, 1743-1750 (2004). |
| |||||||||
All material © John Richard 2003-2004 | Website Developed by Iserloh Design | ||||||||||