|
Phylogenies
2 origins = protein sequence data & cladistics, but we will mostly focus on
DNA sequence data
A simplistic example to illustrate sequence comparisons vs distance data
Consider 3 species: X, Y & Z with 3 genes known to be homologous and identical in
sequence except at 4 base pairs where
ancestral = ATGC and actual evolution was
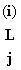
X = ATAA then can tabulate
Y = ACAC the differences
Z = GTGG as shown in table
|
|
X
|
Y
|
Z
|
|
X
|
-
|
2
|
3
|
|
Y
|
2
|
-
|
4
|
|
Z
|
3
|
4
|
-
|
difference method sets up 3 equations in 3 unknowns letting x = distance to
shared point of X; y, of Y; & z, of z then
x + y = 2
x + z = 3
y + z = 4
solving these: x = 0.5; y = 1.5; z = 2.5 & checking solution as bold part of
table get (above)
sequence based method notes best bet ancestral XYZ sequence by
majority rule = ATAV where V = G, A or C
while XY ancestor = AYAJ where Y = T or C & J = A or C
and XZ ancestor = RTRR where R = A or G
and YZ ancestor = RYRM where M = A or G
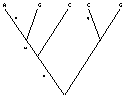
The most parsimonious match is between the XYZ ancestor and the XY so both the
difference method and the sequence parsimony method yield comparable results below
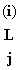 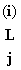
Note that the left (curved) tree is unrooted, but the right (angular) is rooted.
Also that, although the three branch tree is solved exactly, 2 aspects are unrealistic
and one inaccurate:
V must be A, G, C (or less parsimoniously T)
distances of 0.5, 1.5 and 2.5 do not make sense with discrete states (base pairs)
the majority rule assigns A to the 3rd base where G was the "historical" base
Both methods are parsimonious (Occam's razor = KISS)
We know evolution doesn't have to follow parsimony — why do we choose to?
Because deviations are unknown and parsimony is likely to keep error lower than any ad hoc
elaboration
Inaccuracy is built in to parsimony
Miss and often make errors dealing with homoplasy (grow alike - e.g.,
2 separate branches each with A->C) mistaking it for homology (in above example likely
to be grouped together as a single A->C)
Similar problems with reverse mutations (A->C then C->A on the same branch) or
intermediates that will not be seen (A->T->C) or parallel changes that lose the
ancestral sequence (note A vs G for 3rd base above or A->G on one branch vs A->C on
another which leaves us considering one change where two occurred)
The subject
Using figs above as examples, consider these terms from graph theory
graph or network = set of vertices and set of edges with edge = line joining
pair of vertices
vertices = points = real and hypothetical sequences (or loci for distances)
adjacent vertices = those joined by an edge; the edge is incident to the vertices
degree = # of edges incident to a vertex
path = description of route from one vertex to another (can be in terms of intervening
vertices or edges)
connections = paths between vertices; a graph is connected if there is at least one
path between any two vertices
cycle = path that connects a vertex to itself routed thru other vertices but not
repeating any vertex
nodes = vertices but sometimes restricted to internal points (then usually hypothetical
- have degree > 1)
minimally connected = an ideal
roots = deeper connections
dendrogram = a tree = a cladogram (in our studies should be a connected acyclic graph)
OTU = operational taxonomic unit = vertex above but sometimes restricted to terminal
nodes only (have degree one)
HTU = hypothetical taxonomic unit (used when OTU constrained to termini)
binary tree = tree in which all internal nodes have degree < 4; if only one external
node has degree = 2, the tree is full
rooted = tree with special node that imparts direction to it (deepest ancestor)
ordered vs unordered tree = terminal nodes must vs need not be considered in a fixed
order
mathematical theory of networks --> more terms but usually do not
apply here like
loop
# of topologies
unrooted
for 3 points --> 1;
for 4, 3 (look at left side on previous unrooted fig and decide where another sequence
W could be placed - it could divide XY or YZ so that the internal connection keeps WX and
YZ apart or WY and XZ or WZ and XY)
for 5, 5 x 3
for n, (2n-5) (for n > 2)
rooted
for 2, 1
for 3, 3
for 4, 5 x 3
for n, (2n-3) (for n > 1)
Numbers like this soon overwhelm computers
How to root
If an outgroup available, can use it to decide but ultimately not
Usual approach is farthest join from tips but assumes rate constant
Classes of methods
Distance
Historical note - 1st real considerations were Cavalli-Sforza
& Edwards (1967) & Eck &
Dayhoff (1966) + Fitch - Fitch
& Margoliash (1967) + Fitch
(1971).
Assumes:
distances independent
distances from distribution with expectation = sum of values along tree & variance
related to a power p of this expectation
These not necessarily true; can get negative branches so can constrain to � 0 length
UPGMA - like Fitch but non-0 constraint + assumption that lengths relate to an
evolutionary clock. It's based on developments in systematics from
Farris see Kluge
& Farris (1969), Farris
(1970) and Sneath
(1973) - also like Fitch but with non-0 constraint - I don't follow all fine
points of differences
Neighbor-Joining - Saitou
& Nei (1987) - developed the original approach; then it was modified by Studier
& Keppler (1988) - finds pair of OTUs that can be fit with best least squares
deviation (= neighborliness) of predicted matrix first; then creates a mean of these and
repeats procedure until it has completed a tree
Sequence
Parsimony introduced by Wagner and
Camin & Sokal (1965) for systematics - as applied to DNA sequences it's like
illustration given; in systematics some cladists spend an inordinate amount of time trying
to decide which character is ancestral, often applying the constraint that evolution from
an ancestral morph is irreversible. These issues clearly don't apply to bases nor
amino acids. The sequence application has several problems and assumptions below
assumptions: (some claim free of assumption)
Each site evolves independently
Each lineage evolves independently
Ancestral base (or amino acid) unknown for each site
Each individual alteration is a priori improbable
Rates of change along lineages and among sites do not vary so drastically as to make 2
changes in a lineage or site nor likely than 1 in another lineage or site, respectively
(for codons only) synonymous (sense) change is more likely than difference (missense)
problems
Finds only 1 most parsimonious tree; unless algorithm modified if there are more it
doesn't find them
Modeling shows that it doesn't find the historical tree if rates on branches differ
greatly - Felsenstein
(1978)
Modeling also shows doesn't always find historical tree if equal rates or even equal
arbitrarily small rates - Hendy
& Penny (1989)
Parsimony modified by Hendy
& (1982) using branch and bound algorithm to find all most parsimonious trees
Compatibility - based on 2 state concept of Le
Quesne (1969) & subsequent made into algorithm 1st by Estabroook
et al (1976) & later realized similar to Bron &
Kerbrosch (1973) - counts # substitutions for each topology vs minimum from alignment
(max = 3 or 4 if indels count); looks for sites that achieve min and tries to maximize
min. Clearly close to parsimony; assumes:
site & lineage independence as above
ancestral unknown
low rate for most sites as above
few sites very high rates
but which sites high/low not knowable in advance
Evolutionary parsimony - Lake (1987)
{nearly impenetrable} - + invariants - Cavendar
& Felsenstein (1987) -
Lake derives from observation that transitions (A->G, G->A,
T->C & C->T) are more likely than transversions (A->C, A->T, G->C,
G->T & vice-versas) so that transitions = noise when looking for deep relations +
1A C3
\__/
/ \
2A C4 is more likely than
1A A2
\__/
/ \
3C C4 or
1A C3
\__/
/ \
4C A4 because the top requires change only on the middle branch
represent most probable above as [1] (A,A)A,C(C,C) = standard tree
nomenclature then he considers only this + (A,A)A,T(T,T) + (G,G)G,C(C,C) + (G,G)G,T(T,T)
with those like [2] (A,C)A,A(C,A) misleading & [3] (A,C)A,A(T,A) yielding an estimate
of how many are misleading
sounds much harder than it is - looking for [2] -[3] = 0 with least squares
estimate for whether actual result = 0, but only for 'incorrect' trees of the 3 above (=
2nd & 3rd). Very easy to set up on computer; so easy can do it by hand.
Cavendar & Felsenstein broader called invariants because deals with formulas
for expected frequencies of site patterns that should = 0. They consider all 256 4 x 4
patterns instead of restricting to just transversions
Both methods limited by applying only to 4 sequences at a time. Moving these approaches
to >4 has proved intractable so far; Navidi et
al (1991) have begun to solve 5. If there is interest, I can probably track down more
on this kind of approach:
Navidi
et al (1993) gives the clearest mathematical analysis.
Puzzle
Puzzle is a recently released program based on analyzing
quartets of sequences. Recall that at each aligned position
an unrooted tree of four sequences can take one of 3 resolved
forms or a star.Puzzle asks what the most likely result is
comparing over all positions and using a selected weighting
scheme. The result can be visualized as a series of points
located on a triangle (see Strimmer & von Haesler (1997)).
Maximum likelihood - Felsenstein
(1981)
assumes:
site & lineage independence
substitutions have expected rate (can be different for different subs including
transitions)
Based on Bayesian probability
L = P (Data; Hypothesis)
where the semicolon above represents given
example: n coin tosses with odds of heads (H) and tails (T) in question: get HHTHTTTH .
. .. Then with p for H
L = P (HHTHTTTH . . .; p) = pp(1-p)p(1-p)(1-p)(1-p)p . . . = (p^m)*((1-p)^(n-m))
(I)
where m = # heads & n = # tosses
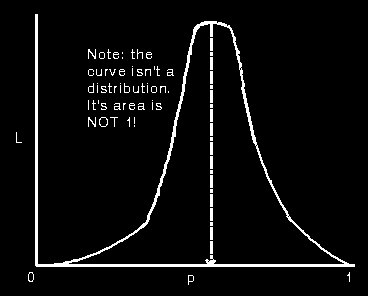
Plotting L vs p to find the MLE of p
A version of Bayes' formula shows for two hypotheses H1 & H2 that
[P(H1;D)] / [P(H2;D)] = [P(H1) / P(H2)] * [P(D;H1) / P(D;H2)] or
posterior odds ratio H1:H2 = prior odds ratio * likelihood ratio. Example
Jury based on previous evidence concludes 1:3 that accused is guilty then gets new
evidence that 15:1 likelihood (based only on new evidence). Then jury's odds should be 15
* 1/3 = 5:1
With trees we want max L so a way to find when tractable is to differentiate L with
respect to p & find where the derivative (slope) = 0. Using eqn I above and noting
that ln L is also max when L is max but that log-likelihood is easier to use so
ln(L) = m * ln(p) + (n-m) * ln(1-p), differentiating
�[ln(L)] / �p = (m/p) - [(n-m) / (1-p)] = 0 for max so
[m * (1-p) - (n-m) * p] / [p * (1-p)] = 0 or m * (1-p) - (n-m) * p = 0 or
m - np = 0
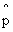 = m/n where p"hat" = m/n where p"hat"
(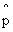 ) means estimate of p ) means estimate of p
with a large amt of data its distribution is roughly normal
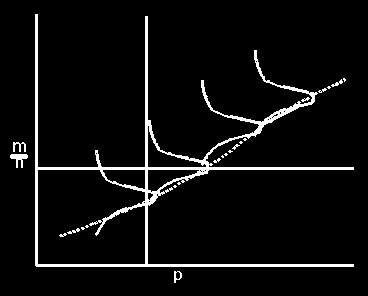
on the ordinate we get the distribution of
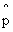 = m/n given p and on the abcissa
we get the likelihood curve for p given m,n. = m/n given p and on the abcissa
we get the likelihood curve for p given m,n.
the std dev of 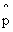 given p can be
obtained from the likelihood curve. Fisher showed that the variance is nearly Gaussian.
The confidence limits when the amount of data is large ~ 1.96 x given p can be
obtained from the likelihood curve. Fisher showed that the variance is nearly Gaussian.
The confidence limits when the amount of data is large ~ 1.96 x s or if use ln(L) within 1.92 of peak where derivative
= 0 and max so
-1.92. Either is 95% confidence interval
Apply this to phylogeny - suppose this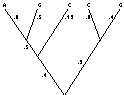
with "branch
lengths" really proportional to expected amounts of evolution. Branch lengths are pseudo times. If one evolves 2x as fast, can pretend 2x as long a time.
Can compute p for these data by assuming
1> lineages start in the same state (with the same base)
2> otherwise they evolve independently
3> events at different states are independent
(Are these assumptions realistic? NO!)
The likelihood for a site on a given tree is the sum of the probabilities of all the
differing ways that pattern could have evolved. More generally
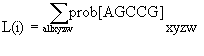
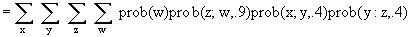
. . .,
with overall likelihood the product
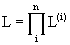
Note: all of the above uses the INDEPENDENCE ASSUMPTIONS
Computational considerations (for those who care; this isn't evolution so will
skip if none care)
likelihood calculated straightforwardly should be very slow, e.g.,
with 10 sequences must sum over 4^9 = 2^18 = 262144 assignments of nts to ancestors. A
cute solution based on Horner's rule for evaluating polynomials economizes by moving
parentheses inwards.
L (i) = 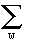 P(w)[ P(w)[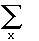 P(x;w)[P(A;x)][P(G;x)]] . .
. this is much faster to compute. One can work outwards from the inside. The pattern of
parentheses follows the shape of the tree P(x;w)[P(A;x)][P(G;x)]] . .
. this is much faster to compute. One can work outwards from the inside. The pattern of
parentheses follows the shape of the tree
the simple algorithm based on this - the pruning algorithm - is
Let 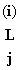 (s) be the prob of everything at site i at or above point j on
the tree, given that j has nt s. (s) be the prob of everything at site i at or above point j on
the tree, given that j has nt s.
At tips if say an A present, then (A) = 1
& (C) = 0, etc for T & G
if we need to consider sequencing error then (A) = 1-e & (C) = e/3, etc.
Going down the tree one can show that if
k l
\ /
j,
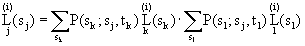
for j=A,T,C,G
This carries out computations exactly
At bottom
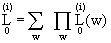
with  = prior prob of w and
L = = prior prob of w and
L = 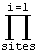
This still leaves huge computational challenges. For a given tree we can get L but we
still must find the best branch lengths for each tree. There are 18 branches in an
unrooted tree with 10 species - thus we have an 18 dimensional optimization to get. Plus
the usual problem that the # of topologies goes up as P (2n-3)
Also need probabilistic model for substitutions
1) Jukes
& Kantor (1969) model
2) Kimura
(1980) 2 parameter model (allows distinction between transitions and transversions)
3) Felsenstein (PHYLIP) generalized 2) for fitting base frequencies found
Some problems with ML
rates at different sites actually different - can try to prespecify to
beat
no allowance for constraints on amino acid or other functional parts of genome
needs known alignment - Thorne et
al (1992) beginning to solve ML alignment problem
ML allows tests comparing hypotheses
e.g.,
a
trifurcation with 4 parameters vs
a
bifurcation with 5 parameters call top likelihood L1 & bottom L2
then 2[ln(L1) - ln(L2)] ~ 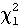
where this is a chi square test with one degree of freedom
butvs
not legit because 0
degrees of freedom
Neither hypothesis is nested in other!
Can argue 1 DF test is conservative
Confidence estimates
So long as stick to nested hypotheses, built into ML
Bootstrapping = a test for the internal consistency of the data - Felsenstein
(1985)
Random sampling of data set with replacement; i.e., draw randomly
n sites from original set of n sites (putting site back into pool after each draw)
then make a set of data with the n sites
use method of choice to make a tree
repeat process until patience or computer time is at a limit
groups that we had prior interest in are significant if they occur in 95% of the
trees
Jacknifing = a similar test; like bootstrap except
no replacement; in more extreme form each sampling is to choose n/2
sites
Paired sites test based on ML extended - Kishino
& Hasegawa (1989)
Given two trees I vs II, look at contribution of each site to ln(L) so
for I
ln(L 1) + ln(L2) + . . . + ln(Ln) & for II
ln(L 1)'+ ln(L2)'+ . . . + ln(Ln)' with differrences
=
ln(L 1) + ln(L2) + . . . + ln(Ln) and can test if ln(Li)
> 0 by a paired t-test or (nonparametric equivalent)
Templeton
(1983) applied it to parsimony by using # steps instead of ln(L) - I'm less certain of
rigour here
Treefile representation - rules and examples from an agreement among Swofford,
Felsenstein, Maddison and others
() parentheses and , commas are used to set off names; any blanks must be
replaced by an underline _; there are limits in length beyond which the name is truncated
(the actual limit varies among programs) so be careful to distinguish all names within the
first 8 characters; a colon : can be used to set off distances after names; the tree ends
with a semicolon ; and is enclosed in parentheses. Also some programs require that trees
be unrooted; to do this the bottom fork will need a 3-way split
Rules to use these
If possible, start at the rooted node
Each time you leave a node traveling away from the root to the leftmost descendant,
make a left parenthesis (
When you leave a node travelling away from the root to a descendant other than the
leftmost, make a comma ,
When you leave the rightmost descendant of an internal node and travel toward the root,
make a right parenthesis )
When you travel to a node for the last time, write the node info (if any - i.e.,
the designation and the distance follwing a colon :
When the tree is complete, make a semicolon ; (at least for use with PHYLIP)
Thus (((A,B),C),(D,E)); =
A
!
!--B
!
!----C
!
!-D
!
E
where the lengths are arbitrary & conversely,
A
!
!- B
!
!- C
!
D
= (A,(B,(C,D))); or with the lengths = n x # of - or !
(A:5,:2(B:3,:2(C:2,D:2))); note that some distances are between internal nodes
As exercises draw the trees for
a - ((A,B)C,E)D;
and write the representation for
b -
For answers see Support materials for lecture 3
| 







