|
|
New Features and History of Changes
Here are some of the more notable new
features and bug fixes since version 1.0. If the previous way Mesquite worked could have
yielded errors in substantive results (e.g., there was a bug in a phylogenetic
calculation) are marked with  . .
Beginning with version 1.1, a list
of new modules in the latest version can be seen by choosing "List
new modules" from the Help menu.
There is also a short list of release
dates.
Version 2.74
New Features
- You can now assign different bin boundaries and colors when using continuous characters in Trace Character History. By default there are 11 bins of fixed widths for the shading of the character states, and the colors are either a pre-defined spectrum or grayscale. Now, you can use Set Number of Bins in the Trace menu to change the number of bins, and Set Bin Boundaries to change bin boundaries. To change colors used, double click on the color rectangles in the legend for Trace Character History and choose the new color. These binnings are specific to a given Tree window. The colors are specific to a given Tree window unless you choose Set Custom Colors as Defaults.
- If you have a tree whose stored branch lengths represent not branch lengths but divergence times, then you can ask Mesquite to reinterpret these numbers so as to assign actual branch lengths, using "Branch Lengths from current, reinterpreted as Divergence Times" in the Tree Window's Alter/Transform Branch Lengths submenu. Thus, if a node had "50.4" as branch length but in fact that was a divergence time, and its immediate ancestral node was assigned "56.7", also a divergence time, then if you used "Branch Lengths from current, reinterpreted as Divergence Times" the node's branch length would be reassigned as 6.3 (= 56.7 - 50.4). This function can be useful if you are importing trees from a program such as TreeAnnotator that assigns divergence times based on BEAST runs. (See item below for a scenario of how this would be used.)
- If you have information attached to the nodes of a tree as specially attached numbers, you can transfer these numbers to become the tree's new branch lengths using Obtain Branch Lengths from Values Attached to Nodes in the Alter/Transform Branch Lengths submenu. This function can be useful if you are importing trees from a program such as TreeAnnotator that places divergence times as comments to nodes in the tree file. (See item below for a scenario of how this would be used.)
- Reading of trees in NEXUS files was enhanced slightly to permit import of information attached to nodes from BEAST and TreeAnnotator. These programs store important information about nodes as comments beginning with "[&". This is not standard NEXUS, insofar as the "[&" comments should include just a single word. You will first need to edit the NEXUS text file by replacing all ampersands to percent signs in the tree description ("[&" to "[%") to permit Mesquite to read the comments. Be careful when you make that replacement: if you change & to % outside the tree descriptions in the TREES blocks, you may render the NEXUS file unreadable). After making this change, read in the tree file. Then open a tree window to show a tree. To check to see if the information was read properly, select the branch info tool (which looks like a question mark) and touch on a branch. Therefore, to obtain branch lengths in the tree derived from TreeAnnotator divergence times, there are three steps:
- Prepare the tree file from TreeAnnotator by replacing & to % in the comments in the tree descriptions, then read the file with Mesquite.
-
In the tree window, show the tree of interest. Select Obtain Branch Lengths from Values Attached to Nodes from the Alter/Transform Branch Lengths submenu, and select the divergence time you want to use (e.g., height or height_median). This will transfer these divergence times into the branch lengths of the tree. The problem with this is that, of course, divergence times are not the same as branch lengths. So, you need to ask Mesquite to recalculate the branch lengths.
- Next, select Branch Lengths from current, reinterpreted as Divergence Times from the Alter/Transform Branch Lengths submenu. This will take the stored branch lengths, intepret them as divergence times, do the appropriate subtractions, and then reassign the branch lengths so as to imply the divergence times. If you now look at the tree with Branches Proportional to Lengths, you will see it is drawn according to those divergence times.
- There is a new menu item in the Tree menu of the tree window, Node-Associated Text, that permits you to display text that is attached to the nodes of a tree. This can be used to display, for instance, text such as the height 95% HPD from TreeAnnotator.
Bug Fixes
- () Protein NEXUS files written by MacClade may be misread by Mesquite 2.73, with some amino acids being coverted to others (specifically, Serine, S, converted to Cysteine, C). This bug only affects version 2.73, and was introduced in an attempt to fix the problem caused by misformatted NEXUS files created by Clustal. More specifically, it is caused by problems in interpreting protein NEXUS files in which there is a SYMBOLS subcommand in the FORMAT command, which is a frequent occurrence with files written by MacClade. [Thanks to Wendy Moore for reporting this.]
- Top BLAST Matches didn't work if used for amino acid sequences; it now works.
- Various small bugs were fixed.
Version 2.73
New Features
- There is a new method to graft subtrees into larger trees, available in the Tree Window under Tree>Alter/Transform Tree>Graft Other Tree. For instance, you could edit a tree of genera, and a separate tree of species within one of the genera, and then graft the second tree to replace the taxon representing the genus in the first. To use this, all of the taxa need to be in the same taxa block (e.g. by using Merge Taxa & Matrices from File). Then, open up a Tree Window showing the receiving tree (e.g. of genera) and a Tree Window showing the subtree (e.g. of species). Go to the receiving tree and select the terminal taxon name (e.g. the genus name) to be replaced by the grafted tree,and choose Graft Other Tree. If you don't select a terminal taxon in the receiving tree, then the other trees is grafted at the base. To work, the subtree and receiving tree cannot share any taxa.
- There is a new file exporter Export Partitions as Separate NEXUS files that writes a separate file for each character group (partition) of a character matrix. This can be useful, for instance to create files to run ModelTest on separate partitions (e.g. 1st, 2nd and 3rd codon positions). You need to first assign the characters to groups by showing the column "Group Membership (Characters)" in the List of Character window and the drop down menu at the top of this column. Then, choose File>Export and select "Export Partitions as Separate NEXUS files".
- Two Taxon Name modifiers have been added: (1) Remove Tokens, which allows you to remove a specified number of words (=tokens) from the start or end of taxon names; (2) Simplify Names, which will simplify names in a standard way for use in MrBayes, RAxML, etc. These are available from the Taxon Names submenu.
- There are now more options for coloring matrix cells by the character state. If Matrix > Color Matrix Cells > Character State is chosen for categorical matrices (other than molecular sequence matrices), then two new options will appear under the Matrix menu: (1) "All Characters Use Same Maximum Value for Cell Coloring" will force Mesquite to use the same color for each state for all characters (e.g., state 1 will always be blue), as opposed to having the state colors vary from character to character as a function of the maximum state for each character; (2) "Set Maximum Value for Cell Coloring" allows you to set the state range to be used for this constant coloring.
- A note in a Notes window now persists when the window is closed; in previous versions of Mesquite it was lost when the window was closed. The note can be seen again by going to Window>Current Windows and selecting the Notes window. To delete a note, you now need to do it explicitly by opening up the Notes window and selecting Delete Note from the window's Note menu.
- You can now export ancestral state reconstructions to a tree description with embedded ancestral states in the format of SIMMAP 1.5, which can then be subsequently read by FigTree. To do this, have the ancestral states recontructed in a Tree window using Trace Character History. Then, in the Trace menu, choose Export Ancestral States Trace>SIMMAP 1.5 Format. The tree file saved can be read by FigTree.
- The default color table for non-DNA and RNA data has been changed so that it is easier to distinguish states visually.
- In importing GenBank files, Mesquite no longer puts GenBank accession numbers as footnotes; instead it stores accession numbers in a special location. GenBank accession numbers can then be viewed in the List of Taxa window by choosing GenBank Number under the Columns menu. It also now uses the DEFINITION tag rather than the ORGANISM tag to obtain the sequence name.
- The Tree Window can be opened in a new mode for editing trees by hand. When requesting a new Tree Window a submenu gives two choices: (1) With Trees from Source, which offers to display trees from a source such as Stored Trees or Simulated Trees, and (2) With Tree To Edit By Hand, which shows a default tree that can be edited and saved into a tree block.
- The "checksum" system for checking the integrity of matrices read from files has been improved to eliminate many of the false warnings given, and to give more diagnostic information if an error is detected.
- Pairwise comparison correlation analysis now supplies text output that can be seen in the text window or harvested using Step Through Trees.
Bug Fixes
- () ClustalX 2, when writing a NEXUS file, inappropriately inserts an unnecessary but problematic SYMBOLS subcommand in the FORMAT command. This is a serious bug in Clustal, because the implication of this subcommand is incorrect according to the NEXUS standard, and should lead to misinterpretation and data corruption. To preserve the data a program should ignore this incorrect subcommand, as MacClade did. Mesquite 2.72 neither ignored the subcommand nor followed the NEXUS standard: it interpreted the subcommand to override the default ACGT symbols list, which caused the character states in the matrix to be mis-read in a different way. As a protection against such files, Mesquite now ignores the SYMBOLS subcommand for DNA and RNA matrices. (This means that technically Mesquite 2.73 is not NEXUS-compliant, but we believe these files misformatted by CLUSTAL would be encountrered much more frequently than DNA matrices with a legitimate SYMBOLS subcommand.) This bug in Clustal has been there for many years, but apparently only was noticed once the files started being read by Mesquite, which previously treated ill-formatted FORMAT commands in a different way than other programs had done. The bug in ClustalX will be fixed in version 2.0.13 (http://www.clustal.org).
- () Export to TNT now writes CCODEs correctly; previously, the characters to which weights, etc., are applied could be in error.
- Various small bugs were fixed.
Version 2.72
New Features
- Parsimony analyses can now be done with meristic data matrices, using Linear Parsimony (analogous to Ordered parsimony with categorical data).
- There is a new tree source, Concatenate Multiple Tree Sources, that brings together several tree sources into one.
- New options have been provided for intepreting how information is attached to nodes and branches of trees, which affects how the information behaves when the tree is rerooted. Branch lengths are treated as if they belong to the branch ancestral to a node, and so when the tree is rerooted, the branch length is reassociated so as to continue to refer to the same branch between nodes. However, the name of a clade needs to remain associated with the same node regardless of rerooting. Ned Young pointed out that other information always behaves as if attached to the nodes (not the branches between nodes). We have changed the behaviour so that branch widths and branch color behave as if attached to the branch. We have also provided a function "Reinterpret Internal Node Labels", available in the Alter/Transform Tree submenu of the Tree menu of a Tree window. When this is selected you can change information associated with the tree as node labels and associate it with the nodes or branches in a different form. This can be useful, for instance, if you have imported a tree from MrBayes with posterior probabilities attached as node labels. Being attached as node labels, the probabilities will be mis-associated with nodes if the tree is rerooted. You can move the probabilities from node labels to numbers attached to branches using Reinterpret Internal Node Labels.
- The reporting of crashes and checksum errors has been improved.
Bug Fixes
- () When taxa were deleted, there was a chance that a crash would occur, and this crash could corrupt data. This crash occurred inconsistently, but if it did occur, then you would have received a warning that data might be corrupted. If you ignored the warning, then your character data or other information could have become associated with the wrong taxa. This bug has been fixed.
- () When a 3 dimensional (i.e. multi-item) continuous character matrix was exported to a file, and in a few other contexts, the exported matrix would have the items written incorrectly. This did not affect typical continuous matrices, only those with multiple items in each cell of the matrix. This bug has been fixed.
- Reading and writing of DNA matrices was fixed so that it preserved lower case symbols
- Trees exported to PHYLIP and other formats are now written without "decorations" such as branch colors, which tended to render the trees unreadable by other programs
- Sorting of columns now respects numerical order (and thus 2.0 is sorted as between 1.0 and 10.0)
- Various small bugs were fixed.
Version 2.71
New Features
- The crash reporting system now invites you to enter an email address so that the Mesquite developers can contact you if we need more information to diagnose or fix a problem. Entering an email address is optional.
- A new scripting command, getValuesTabbed, can be sent to chart windows to obtain the chart values as a simple list of numbers separated by tabs.
Bug Fixes
- Various small bugs were fixed.
Version 2.7
New Features
- There is a new crash reporting system that can send reports to the Mesquite development team. When certain crashes occur, a dialog box will appear offering to send a report. We urge you to permit the report to be sent; it will help us fix bugs.
- Speed and memory use has been improved considerably for large matrices and trees. This permits many more taxa and characters to be conveniently handled.
- Consistency Index and Retention Index can be calculated for trees with individual characters and with matrices. These are available in various places, for instance in the List of Characters Window, Column>Number for Character>Character Value with current tree...
- NEXUS tree files without translation tables can now be read directly, without first reading or making the corresponding taxa block.
- The BiSSE simulation of trees and characters (available through Trees & Diversification Characters in the Taxa & Trees menu) now asks in the simulating dialog box what prior probability to put on state 1 at the root. The default is to use the equilibrium (stationary) frequencies as the prior, but you can enter 0.0 to indicate the state at the root is necessarily 0, 1.0 to indicate it is necessarily 1.
- Tree simulations normally are set to be repeatable; i.e. if you ask for tree 1 several times, you will get the same tree 1. This can now be turned off by deselecting the menu item Simulation Repeatable. This could be useful in scripting under different scenarios to ensure each scenario uses an independent random number sequence.
- There is a new tree window tool, Node Age Constraints (shaped like an anchor), that permits age constraints to be assigned to nodes. These constraints are not yet used by any standard Mesquite module, but could be used by optional modules. More details are given here.
- The function to save trees to a file without holding them in memory, previously available under File>Export as "Export NEXUS Tree File from Tree Source" has been moved to Taxa&Trees>Save Trees To File from.
- A new source of trees is Concatenate 2 Tree Sources, which supplies trees from two separate sources as if they were one source. Trees from the first source have an attached value of "1", while trees from the second source have an attached value of "2". Concatenating tree sources can be useful for example to concatenate two tree files that are too large to read into memory at once. More details are given here.
- There is a Revert To Saved menu item in the File menu that reverts the project to its last saved state. Choosing this menu item is the equivalent of closing the file without saving, and then rereading it from disk. If you have linked files open, then choosing it is equivalent to closing all files in the project without saving, and then rereading the home file of the project from disk.
- There is a new submenu, "Tree Inference", in the Taxa&Trees menu, that shows functions to infer phylogenetic trees. Currently the only built-in tree inference methods are Mesquite's heuristic Add & Rearrange, and simple Cluster Analysis. However, we anticipate that optional packages will soon be released to add functions.
- The Tree Window has a new scripting command, getAsTable, that returns a string in the form of a table that can be converted into a tab delimited table. It summarizes ongoing calculations on the tree such as traced characters and values at nodes. The format is "Calculation Type<tab>node value<tab>node value<tab>...".
- Character matrices can be marked as "hidden", which means that they will not be included in most menus and dialog boxes in which you can choose matrices, and they will not appear in the project panel at the left of the window. To set the visibility, go to the List of Character Matrices, Column>Visibility of Matrix. Once the column appears, select the rows for the matrices whose visiblity you want to change, and choose Toggle Visibility from the drop down menu at the top of the column.
- You can now suppress decorations on taxon names in the tree window (e.g. the "*" that indicates a footnote is available) by deselecting Show Footnotes Etc. under Taxon Names in the Drawing window.
- The tree window now has a menu item, Legends to Default Positions, that can recover legends that have become lost off screen.
- The Square Tree drawing form now has a Corners submenu that permits drawing with rounded corners:
- You can assign a name to your project other than the name of the home file. The project name is then displayed on the window's title bar. To change the name of the project, touch on "Project" near the top of the project panel at the left of the window. In the drop down menu, choose the menu item Name of Project...
- When a duplicate taxa block is read, for instance in a linked file, the first taxa block is not reordered. (Actually, it is reordered temporarily to complete reading of the linked file, but then reverted to the original order.)
- Mesquite now requests more memory by default.
- Mesquite's phone home system now sends the following information to the Mesquite server when you start up Mesquite: your IP address, your operating system, and your version of Java. (Similar information is sent to any web server you visit when you browse the web.) This is sent so that we can better prioritize what operating systems and Java versions to support. If you want to turn this reporting off, deselect "Check for Notices on Mesquite Server" from the Defaults submenu of the File menu.
- With a character matrix open, you can now quickly remove
all invariant characters by choosing Remove Invariant
Characters from the Alter/Transform
menu of the Matrix menu.
- The Merge Taxa command now allows you some control over how
the name of the merged taxon is formed.
Bug Fixes
- () There was a bug that could scramble the Taxa Associations used to indicate how taxa in one taxa block are associated with taxa in another block (e.g., for genes in species). Scrambling could occur if taxa were reordered then new taxa added, and would result in the wrong taxa being listed as associated. This bug has been fixed.
- () There was a bug that could place incorrect data into a continuous valued character matrix if a section was cut, and then Undo called. The error would have likely been obvious. This bug has been fixed.
- () There was a bug that caused Mesquite to misinterpret typed character states when editing a character matrix that used certain non-standard lists of symbols. This would have generated a warning about checksum errors. This bug has been fixed.
- ()
The Translate DNA to Protein feature had a bug in that it on occasion removed the last amino acid; this has been fixed.
- Trace Character over Trees could sometimes display the incorrect character name when characters were excluded. This bug has been fixed.
- The BiSSE viewer would record the wrong character number into a file when saved, so that when reopened the file would disply statistics based on a different character than it had previously. This is fixed.
- We have fixed a bug that would cause Mesquite to issue an unnecessary warning that the log file could not be written.
- There was a bug in the Undo system when a cell of a character matrix was being edited; repeated requests to Undo could place unexpected states into the cell. This is fixed.
- Some bugs with tree window scrolling, introduced in version 2.6, have been fixed.
- Various other small bugs were fixed.
Version 2.6
New Features
- There is a new interface simplification system.
- There is a new system for installing updates for Mesquite and extra packages. If an update is available, you will be notified when Mesquite starts up. Updates that you choose not to install will still be available via File>Available to Install or Update.
- The Tree Window's automatic sizing of the tree drawing has been improved, and the menu items controlling tree sizing have changed.
- You can now display boolean values for characters in the information strip at the top of the data matrix,
using the Matrix>Add Info Strip>Boolean Info Strip submenu. The two initial booleans whose values you can
display in this way are whether or not the character has two or more states, and whether or not it is included.
You can remove the information strip by choosing Remove Info Strip in the menu you get when you touch on the info strip.
- You can now export distance matrices from various sources (patristic distances, pairwise sequence distances, geographic distances) to a text file. This can be done by selecting Export from the File menu, and choosing "Export Taxa Distance Matrix".
- Mesquite can now read treefiles produced by BEST (Bayesian
Estimation of the Species Tree), the program by Liang Liu.
- The menu items for Summarize State Changes Over Trees and Summarize Changes In Selected Clade have been moved to the Analysis menu
- The "phone home" system by which Mesquite checks its website to notify you of updates now tells the server which version of Mesquite you are using. Mesquite does not transmit any other data about you, except what your web browser does when you go to a website. (You can disable the "phone home" system in the Defaults submenu of the Edit menu.)
Bug Fixes
- ()
If consensus trees were constructed over trees with branch
lengths stored, the average branch lengths of the terminal
branches of the consensus tree was incorrect. This has been corrected.
- ()
If new characters or taxa were inserted in an existing matrix or taxa block anywhere but in last position, then subsequent characters or taxa may have had their status in selection sets corrupted. This would not directly lead to a substantive error, but could indirectly if those selection sets were subsequently used for manipulating data. This has been fixed.
- ()
If new characters or taxa were inserted in an existing matrix,
for example by importing FASTA files, then subsequent use of
the manual alignment tools (e.g., the multiple sequence splitter)
might in rare circumstances cause loss of a few bases from
the ends of sequences. As far as we know, Mesquite would have given
a warning if this happened. (Thanks
to Jolanta Miadlikowksa for reporting this.)
- Mesquite can now more successfully import and export PHYLIP files.
- Undoing some operations in the data matrix caused Mesquite to crash.
This has been fixed, but only by preventing the ability to
undo some actions that could lead to changes in the entire
matrix if the matrix is very large. You can set the maximum
size of the matrix that is fully undoable using
File>Defaults>Matrix Limits for Undo...
- INTERLEAVED NEXUS files with more than 2000 characters per
interleaved block were written incorrectly by Mesquite; this has been fixed. (Thanks
to Jolanta Miadlikowksa and Johan Nylander for reporting this.)
- Moving multiple sequences in Mesquite using the "Splits multiple
sequences" tool was very slow; this has been fixed.
- Shift to Match Other was not finding many sequences that
matched that were matches; it is now much improved.
- Missing data in FASTA files is now correctly written.
- Various other small bugs were fixed
Version 2.5
New Features
- The default appearance of Mesquite's windows have changed.
You can alter the general colors used by choosing items from
the File>Defaults>Color Theme menu.
- Consensus tree calculations
are now available in the standard Mesquite packages (there
are separate consensus tree calculations in the TreeSetViz
package). Strict, semistrict, and majority rules consensus
are available.
- Results of calculations over multiple trees in the Tree Window are available without scripting through the new Step through Trees feature.
- Mesquite's parsimony ancestral
state reconstructions can now examine individual Most
Parsimonious Reconstructions (MPRs), and calculate
how many there are.
- There are two Summarize
State Changes Over Trees modes, both
of which summarize the frequencies of each sort of character
change (e.g., from state 0 to state 1) based upon character
histories reconstructed over each of a collection of
trees.
- There is a new Project Panel at the left of most windows to help you keep track of your data objects such as taxa blocks, character matrices and tree blocks. With it you can display, export, rename, or delete these objects.
- The Tree Window has a new Tree
Info Panel, available by touching the blue "i" (
 )
in the tree window. This panel shows basic information
about the tree and its notes. It also has a Values section
in which you can display values from various calculations
(character likelihood, etc.). )
in the tree window. This panel shows basic information
about the tree and its notes. It also has a Values section
in which you can display values from various calculations
(character likelihood, etc.).
- Selection of taxa and branches in the Tree Window has been changed to be more natural, using the standard arrow cursor via clicking and dragging.
- The Character Matrix editor has various improvements:
- There is a Matrix Info Panel, available by touching the blue "i" () in the matrix editor window. This panel shows basic information about the matrix and its entries. For categorical matrices, you can edit character names and character state names; for sequence matrices you can edit a comment for the particular esquence; for continuous matrices you can adjust the matrix's items and their names.
- Copy/paste of molecular sequences have been improved.
- Character names can be written diagonally for categorical and continuous matrices.
- You can now change the symbols
used for missing data and inapplicable (gaps) using Matrix>Current
Matrix>Missing Data Symbol and Matrix>Current Matrix>Inapplicable
Symbol
- For DNA or RNA sequence data, there is a new feature
to alter data (available in the Matrix>Alter/Tranform>Convert
to RY that converts A or G to R, and C or T to Y.
- There are two new tools for manual
sequence alignment. These are described in the Align manual:
- Selected Block Mover
- MultiBlock Splitter
- There is a new data type, meristic,
for counts (number of teeth, etc.). This is not widely supported
in analyses, and has been introduced now for several internal
bookkeeping functions.
- The boxes for taxon names and character names in the Character Matrix Editor are now colored independently from the internal cells of the matrix. These have their own submenus in the Matrix menu to control them.
- The Tree Legend has been renamed as "Values for Current Tree". Its functions are also duplicated in the Tree Info Panel "values" section.
- By default menu items and dialog boxes show all choices, not just the primary choices.
- The tree drawing menu has been reorganized to put Taxon Names Angle under Names.
- Select taxa through search of taxon
names. This feature allows you to select taxa if a portion
of their names match the specified text. It is available
from the Select>Select Taxa submenu in the character matrix
editer, or in List>Select Taxa submenu in the List of Taxa
window.
- There is a new module to calculate the average tree depth,
that is, the average length of the paths between terminal taxa
and the root, called "Average Tree Depth".
- Export NEXUS Tree
File from Tree Source. This feature allows
you to save a text file directly from a tree source.
Bug Fixes
- () The correlation analysis using pairwise comparisons occasionally would display results that were not complete (not based on a full sample of pairings).
- () When taxa were added, deleted or reordered, some special information such as branch widths, colors and notes associated with the nodes of trees became associated to the wrong nodes. Branch lengths were not affected by this.
- () Simulations of tree and character evolution simultaneously using "BiSSE Trees & Characters" (available via Trees & Diversification Characters menu item in Taxa & Trees) in some circumstances would have been done using parameters different from those specified in the dialog box, depending on the sequence with which parameters were specified.
- Hennig86/NONA file export had incorrectly
counted comments. Hennig86/NONA file export now writes CCODE
specifications explicitly, not assuming a default.
- The autotab feature to the quick key selector tool now works
correctly in the data editor.
- Many other smaller bugs were fixed.
Version 2.01
New Features
- "Phone home" system. Mesquite now checks its website on startup to see if there are notices for users, for instance about updates or bugs. (This can be disabled in the Defaults submenu of the Edit menu.) Other packages can also be set to check their websites. This checking does not transmit data about you other than what your web browser does when you go to a website.
- The search facility to help you find features ("Search Features", available in each window:
 ) has been improved to include menu items and tools in open windows. ) has been improved to include menu items and tools in open windows.
- To help minimize user confusion, there are new options in how Mesquite by default presents choices of modules to the user. First, dialog boxes that offer a choice of module may now show more detailed choices. For instance, whereas previously you would be offered a choice "Tree value using character", now there will also be a list beneath it of the particular values available:
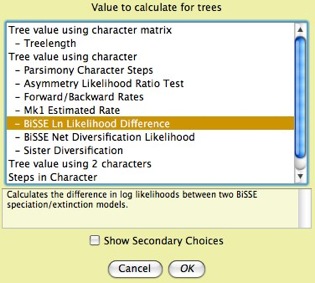
This new default can be reverted to the old style by deselecting "Show Subchoices in Module Dialogs" in the Defaults submenu. Second, calculations that use characters or matrices can be set to use Stored Characters and Stored Matrices by default, without asking. This default may help novice users who otherwise would be confused by a request about simulations and so on. You can set this default as explained on the Characters page.
- The Defaults menu is now a submenu of the File menu, and is available from all windows.
- GenBank and GenPept import. Mesquite can
now import GenBank and GenPept files. If multiple files are dropped
onto a molecular matrix of the same sort (e.g., DNA), then
these sequences will be added to the matrix.
- Drag and drop of sequence files. You can now drag a collection of FASTA, NBRF,
or GenBank files and drop them onto an existing molecular matrix.
Mesquite will ask you to specify the file format, and then
it will read in the sequences and append them to the bottom
of hte matrix
- New alignment tools (in the Align package):
- Align DNA to Protein. A DNA matrix can be aligned to match that of a translated amino acid alignment.
- Muscle Align. A portion of a sequence matrix can be sent to MUSCLE to be aligned and then reimported back into the matrix.
- Shift to Minimize Stop Codons. Sequences will individually be shifted 0, 1, or 2 based to minimize the number of stop codons
in each sequence. This is useful to set the reading frame of each sequence, for example for subsequent translation to amino acids.
- Tree of Life Web Project linking and other online databases. Trees from the Tree of Life Web Project can
be downloaded and navigated. In addition, a new tool in the tree window will search for taxon names in various on-line databases, including TreeBase, iSpecies, etc.
- Export NEXUS Tree File. This new exporter allows you to save a tree file (optionally including a taxa block) from a Mesquite project.
- A Search Within Clade Tool is available in the
tree window. This tool rearranges the branches above the node
touched looking for a better tree.
The objective function used (e.g., treelength, or something
else) can be chosen by the user. This is similar to MacClade's
search tool in that it directly rearranges the current tree.
- Randomly Resolve Polytomies has a new convention. In previous versions it paid little attention to branch lengths, simply using the same convention as when a user moves branches. This resulted in the descendant nodes acquiring different distances from the root than they had had before resolution. The new convention is to preserve distance from root when resolving polytomies. The user however must specify what portion of branch lengths to put on new branches formed during the resolution.
- The Contained Associates tree drawer, most commonly used to display gene trees within species trees, has been renamed as Contained Gene (or Other) Trees.
- The Defaults menu of the Log window has been moved to a submenu of the File menu, and is now available in all windows.
Bug Fixes
- Various bugs concerning behavior of windows and dialogs under Linux and Mac OS X were fixed.
- () Deep coalescences were miscounted if a species or population lacked any containing genes. Such a species or population would contribute -1 to the deep coalescence count in such a circumstance (it should have contributed 0). This could, for instance, result in a deep coalescence score of a negative number, but it could also simply lower the total. This bug did not affect relative scores among trees (as long as they contained the same set of populations and genes) and so many analyses, such as searching for optimal species trees, would be unaffected. This bug was fixed in build j28 of version 2.01. The very first build of 2.01, j27, released for a few hours, has this bug.
- () The Number of Stops in a taxon in some
circumstances was underestimated if there were some characters whose codon positions were not specified.
Version 2.0
New Features
- Search system — A "Search"
text entry box is now available at the top of most windows (in
the Log window, at the bottom). Enter text there and hit Return,
and the text will be sought. There are several modes, marked
by different symbols:
- & — Search Features, AND:
searches through information about Mesquite's features and
returns a description of those features matching ALL of
the search terms
- / — Search Features, OR: searches
through information about Mesquite's features and returns
a description of those features matching ANY of the search
terms
- d — Search Data: searches the current
data file or project and returns objects (e.g. taxa, characters,
sequences) that match the search string.
You can choose the mode by touching on the symbol until the
desired mode appears. In the Search Features modes, the results
also give indication as to how the feature might be accessed.
(There may even be a Try It! link that offers
to invoke the feature.)
- New windowing system — By default, all of the windows of a single project now
appear as tabbed panels within a single window. This change was
made to limit window proliferation. If you want to "pop out"
a tab to be a separate window, hit the curved arrow button within
the tab (you can "pop in" the window again using the
curved arrow button at the upper left side of the separated window).
You can revert to the old system of many windows using the Open
Windows as Tabs item in the Defaults submenu of the File menu.
- Wizard-style dialogs — In many contexts
Mesquite needs to ask a series of questions when a feature is
requested. In previous versions these would appear as a series
of dialog boxes; in version 2 these now appear as a single dialog
that moves from one question to another, in the style of a wizard.
This change was made to make an area in the dialog to give a
more thorough explanation of the request, and to minimize the
visual distraction of changing dialog sizes. You can revert
to the old style of separate dialog boxes (which may work better
on some operating systems) using the Use Wizard-style Dialogs
item of the Defaults submenu of the File menu.
- Diverse package — There is a new package
of modules that estimates and simulates models of speciation
and extinction. Some of its features are:
- Estimating speciation/extinction rates
- Detecting state-dependent differential speciation/extinction
(BiSSE)
- Birth/Death tree simulation
- Simulations with evolving characters that affect speciation/extinction
- Lineages through time plots
- Coalescence simulation with migration —
Coalescence within a population/species tree can now be simulated
with a selected rate of migration per generation among populations.
- Trace Character Over Trees — This feature
has been improved in several ways. The fraction of trees without
a node is now shown, as is the fraction of trees with the node
but not counted as they have ambiguous state assignment (by
whatever criteria are chosen). The states at the terminal taxa
are now shown. There is a new calculation mode, Average Frequencies
Across Trees, available if the ancestral state reconstruction
module yields frequencies or probabilities; this calculates,
for each state, the average frequency across all trees (including
trees in which the frequency for that state is very low or zero).
- Sample Trees Directly from File (now called Sample Trees from Separate File) — One
can choose as a source of trees a random subsample of the trees
contained within a treefile.
- Undo — You can now undo some changes
you might make to the character matrix in the character matrix
window. In particular, edits of an individual cell, or changes
made by the items in the Matrix>Alter/Transform menu, are
now undoable. Other changes (e.g., deleting a character) are
not yet undoable.
- Consensus sequences — One or more consensus
sequences can be displayed in the character matrix editor.
- Adding sequences to an existing matrix —
FASTA or NBRF files dropped onto a matrix of DNA or protein
sequences can be read in and appended to those matrices.
- Fusing genes to single matrix — In
concatenating matrices, you can now contatenate matrices with
different sets of taxa, facilitating fusion of different genes
into a single matrix
- Alternative naming schemes for taxa —
This feature was introduced in version 1.1 as "Archived
names". It has been expanded to permit multiple archived
naming schemes.
- Matrix coloring to aid manual sequence alignment
— There are two new Color Cells options that aid with
manual alignment. One ("Aligning Colors") uses colors
that emphasize purine-pyrimidine differences; the other highlights
sections that appear to be slightly misaligned.
- Various other smaller improvements have been made, for instance
you can now import a PHYLIP tree file directly (without having
to establish a taxa block first), and you can choose darker
colors when assigning colors to objects.
Bug Fixes
- ()
There was a bug in writing decimal numbers as strings. Frequently,
the 7th or 8th significant decimal place of a number would be
written incorrectly. This did not affect how data were stored
in the file; it affected only the reporting of results and screen
display. This is unlikely to have a serious effect on published
results for two reasons. First, it usually affected only the
7th or 8th significant decimal place. Second, the situations
in which it affected a more important decimal place were obscure
and perhaps rarely used by users. These are: Density output
in Scattergram; Parameters Explorer for newer likelihood calculations;
Location of changes in Stochastic Character Mapping. In these
cases, the 3rd or 4th decimal place could be affected. This
bug has been fixed.
- ()
On Windows operating systems, pasting into the data matrix often
resulted in the loss of the first character of some rows. This
may have been benign, but occasionally would result in data
being pasted incorrectly (e.g. 1.234 received as 0.234). This
bug has been fixed.
- ()
Among the coalescence calculations, the method to resolve a
polytomous contained gene tree failed to resolve all polytomies
needed, leaving some unresolved. This resulted in inflated Deep
Coalescence counts. This bug has been fixed.
- ()
Fixed bug in which parsimony step matrix would be incorrectly
written and could not be re-read properly if it refered to characters
with more than 10 states.
- Fixed bug in which Recode dialog box would disappear unexpectedly.
Version 1.12
Bug Fixes
- ()
In versions 1.1 and 1.11 of Mesquite several of the simulations
and randomizations had a bug in choosing the random number seed
for the first item simulated or the first randomization. The
designed behavior is for the simulations to begin with a random
number seed based on the current clock, and then from that calculate
base seeds for each of the replicates using the procedure described
below under version 1.1 bug fixes. Version 1.12 has been corrected
to use this designed behavior. However, in 1.1 and 1.11 for
several of the simulations the first replicate always used 1
as its random number seed, although all subsequent replicates
used the correct method as designed. This bug did not affect
independence of replicates within a simulation of multiple replicates
(except when multiple tree blocks were simulated using "Simulated
Tree Blocks"), but it did affect independence among separate
simulations/randomizations. Since the bug affected only the
first replicate, it would not be expected to affect significance
values unless very few replicates were used. The affected simulations/randomizations
are: Simulated Trees (first tree used seed 1 always); Simulated
Tree Blocks (first tree of each block used seed 1 always); Randomly
Modify Trees (first tree used seed 1 always); Randomly Modify
Matrices (first matrix used seed 1 always).
- ()
When taxa are selected by the user and Rarefy Tree is used to
generate trees with a requested number of taxa randomly deleted,
taxa are chosen for deletion randomly only from among those
currently selected. In previous versions of Mesquite this did
not work correctly, in that frequently fewer than the requested
number of taxa were deleted. This may have been evident to the
user because the modified tree would have contained more taxa
than expected. This bug is fixed in 1.12.
- ()
The likelihood calculations for categorical characters were
designed only for characters with a contiguous series of states
starting at 0, and to prohibit other characters. That is, a
character with states 0, 1, 2 would be permitted, but a character
with states 1 and 2, or a character with states 0 and 2, would
not be permitted. This requirement was mentioned in the manual,
but the requirement was not enforced properly within the program,
and thus Mesquite permitted the calculations with some such
characters. The reconstructions are incorrect. In version 1.12
the prohibition is correctly in place; Mesquite will refuse
to calculate likelihoods with such characters.
- ()
A single character matrix can be modified from various windows,
such as the List of Characters window (e.g., by moving, deleting
characters) and the Character Matrix Editor. When a character
matrix editor is open and a cell is being edited, the editing
field could sometimes be misapplied to the wrong character or
taxon if elsewhere characters or taxa are deleted, added or
changed in order. The effect of this is that the state of one
character or taxon can get copied inadvertantly to another character
or taxon. This bug has been fixed.
- A bug in the shading of character
states for Trace Character History for continuous characters
has been fixed. In previous versions there were situations in
which a branch was given a color slightly offset from its correct
color according to the legend and its state value. (The correct
state could always be seen, however, by holding the cursor over
the branch.) This has been fixed.
- Fixed bug in Find Sequence (Matching
sequence).
Version 1.11
Bug Fixes
- ()
In previous versions of Mesquite, a Taxa Association between
taxa blocks, for example one which records what haplotypes belong
to what species, could come to have incorrect linkings if taxa
were reordered in the file. This is not directly a substantive
bug, but could yield calculations with errors if the user does
not notice that taxa have become mis-associated. This bug has
been fixed
- Clustal Align now works under Windows
- Various other small bugs were fixed.
Version 1.1
18 May 2006
New Features
- Character evolution
- Gene tree/species trees
- Alignment (see
the
Align Package manual)
- Manual alignment tools
in editor (Block Mover, Sequence Pusher)
- Automated pairwise alignment
tool in editor
- Submission of a selected region
in the matrix to be aligned by ClustalW and
then reincorporation of the newly aligned region into the
matrix
- Genetic
Codes
- Assignment of genetic
codes to individual characters
- Translation
of nucleotide data to protein data
- Molecular Data
- Distances
- Jukes-Cantor, Felsenstein 81,
Kimura 2-Parameter, Felsenstein 84 distances
- Great Circle distances in the
separately-released Cartographer
package
- Charts
- Values for Taxon Pairs now available
in Bar & Line Charts and Scattergrams, allowing one to
plot, for example, uncorrected distance versus corrected distance
for all taxon pairs within a matrix.
- Line charts
- Bar & Line charts can now
show cumulative results in various fashions
(e.g., cumulative, cumulative average, reverse cumulative
average)
- Interactions
with GenBank
- Import into matrix the top BLAST
matches to sequences selected in matrix
- Interaction with MrBayes
- live updating
of trees produced by ongoing MrBayes analysis
- exporter that merges
matrices and includes a MrBayes block summarizing the
partitions in the resulting matrix. Merged matrix can be mixed
(DNA, protein, and morphological) using MrBayes's modified
NEXUS format.
- Managing matrices and files
- fuse
matrices into a single matrix, even if matrices
refer to different taxon names (e.g., sequences of different
genes), using a table
of correspondences among taxa blocks
- alternative
taxon names (permitting you to have alternative naming
schemes for taxa in a file)
- include
taxa from NEXUS, NBRF or FASTA files into current file,
e.g. to add newly acquired sequences to an existing matrix
- import/export Phylip tree files
- Randomization (reshuffling) of matrices
can now respect taxa partitions; also can reshuffle within taxa
- PDF file saving
of graphics of trees and other windows
- Colors of character tracings can
now be changed
- Simplified installation process
for Windows computers
- A complete
list of standard modules has been added to the manual.
Bug Fixes
- ()
In previous versions of Mesquite, simulations and randomizations
of trees and characters used a shortcut to go to the n'th replicate,
cycling the (pseudo-)random number generator n times before
entering the replicate. This caused adjacent replicates to be
not entirely independent. In most cases we expect this bug would
not have had a biasing effect if sample sizes were large. This
bug has been fixed by adding 1 to the random number seed after
cycling n times to generate independent random number sequences
for each replicate.
- Various bugs and inefficiencies
affect memory use have been solved in file reading, tree display
and printing
- There were some copy/paste bugs,
especially under Windows, that have been fixed
- Swapping in heuristic tree search
now retains only unique trees
- NEXUS files with high-ASCII characters
(e.g., letters with accents) are better handled
- Various other minor bugs and inefficiencies
have been fixed
- build h61 (released 23 May 2006)
has a few extra bugs fixed: (1) Categorical likelihood calculations
no longer complain if root has zero length; (2) Bug in Java
1.5 on Mac OS X caused list dialogs not to respond properly
to clicking "Show Secondary Choices"; workaround implemented;
(3) Incompatibility with PDAP file exporter solved; (4) directory
choice dialog under Mac OS X improved.
Version 1.06
New Features
- The window showing annotatons for
taxa, characters and cells of a matrix has been integrated into
the Character Matrix Editor as a panel within that window. This
makes it easier to annotate without having to manipulate two
windows. This panel can be requested by the Show Annotations
Panel item in the Matrix menu, or by touching on the
small button (
 )
near the lower left of the Matrix Editor (beneath the taxon
names). The pencil tool ( )
near the lower left of the Matrix Editor (beneath the taxon
names). The pencil tool ( )
has disappeared. Another change is that images in annotations
can now be remote (i.e. URL's). Also, the annotations panel
is also available in the List of Taxa window (for annotating
taxa) and the List of Characters window (for annotating characters).
In the list windows, the panel can be requested by touching
on the small button ()
at lower left of the table. )
has disappeared. Another change is that images in annotations
can now be remote (i.e. URL's). Also, the annotations panel
is also available in the List of Taxa window (for annotating
taxa) and the List of Characters window (for annotating characters).
In the list windows, the panel can be requested by touching
on the small button ()
at lower left of the table.
- Annotations can now be attached
to the states of a categorical character, within the State Names
Editor (available from the Matrix menu of the Character Matrix
Editor). The annotations can be viewed by choosing Show
State Annotations in the State_Names menu or by touching
on the small button at lower left of the table ().
- A legend is now available for the
colors in a Character Matrix Editor. The legend can be shown
by selecting Show Color Legend in the Matrix
menu, or by touching the small button(
 )
at lower left of the Matrix Editor (beneath the taxon names).
If you double click on a color in the matrix, the editor will
move to a cell with that color. )
at lower left of the Matrix Editor (beneath the taxon names).
If you double click on a color in the matrix, the editor will
move to a cell with that color.
- There are buttons at the lower left
of the Character Matrix Editor to open the List of Characters
window (
 )
and the List of Taxa window ( )
and the List of Taxa window ( ).
Reciprocally, the List of Characters window has a button ( ).
Reciprocally, the List of Characters window has a button ( )
to show the Character Matrix Editor. )
to show the Character Matrix Editor.
- Character matrices can now be viewed
in a "Birds-eye view" with very narrow
columns.
- There is a State Names Strip
available for categorical matrices under the Matrix menu. This
shows a strip along the bottom of the matrix in which the names
of characters and states are shown. Similarly when Bird's Eye
View is used for a matrix, a Bird's Eye Closeup Strip
appears below the matrix so that you can more clearly see the
states of the characters of focus. Both of these strips can
be made higher by going to their upper edges, clicking and dragging
upward.
- You can now view a character matrix
in more than one window, by requesting an Extra Matrix
Editor from the Characters menu. This is useful, for
instance, if you want to have the cells of the two matrices
colored differently.
- The order of characters can now
be managed through stored character orders. A Default
Order of characters is automatically stored as characters
are created. This default order is visible in the List of Characters
window as the first column. If the order of characters is accidentally
or intentionally changed (for instance using the Sort
Tool) then the default order can be recovered easily
by applying the Sort Tool to the Default Order column of the
List of Characters window. In addition, you can store and reload
other character orderings by showing the Stored Character
Order in the List of Characters window (Columns menu).
You can store the current ordering and recover previously stored
orderings using the drop down menu from the Stored Order column
heading. When you do this, you will see the numbers in the column
change, but the characters will not immediately be reordered.
The reason for this is that what is stored and displayed is
simply a number for each character marking its position. It
is up to you to use that ordering to resort the characters.
Thus, to recover a former sequence of characters, load the stored
ordering, then touch on its column with the sort tool.
- There are new facilities
for searching for sequences in molecular data
files. Thes are found in the Edit menu for editor windows of
molecular sequence data. These are:
- Copy
Sequence (at bottom of Edit menu): This copies
the selected cells of the matrix into the computer's clipboard
as a sequence. That is, whereas the standard Copy would
place into the clipboard selected pieces of the matrix in
tab-delimited text format (e.g., if the sequence AATCA is
selected, "A-tab-A-tab-T-tab-C-tab-A" would be
copied), this modified Copy Sequence command does not include
tabs (thus, "AATCA" would be copied). This style
of copying is useful when interacting with programs like
Sequencher (TM).
For instance, if you want to find a piece of sequence in
a matrix in Mesquite within a chromatogram viewer of Sequencher,
do the following: select the sequence in Mesquite, choose
Copy Sequence, then go to Sequencher, select Find Bases,
and paste the sequence as the search string.
- Maintain
Target Match (in the Find Sequence submenu of the
Edit menu): This highlights and maintains highlighted the
first occurence of a given sequence in a given taxon. First,
you are asked which taxon to search in. Then, it displays
a panel like this:

underneath the matrix. The first button (red X) is to close
the panel; the second pauses the search; the third allows
you to select another taxon as your focus. If you type a
sequence into the text area, the matching sequence (if any)
will be highlighted in the matrix. Mesquite is constantly
monitoring this text, and so you don't need to give any
command to find again if you change the text. This is useful
if working with a program like Sequencher. If you see a
stretch of sequence while viewing chromatograms that you'd
like to find in the matrix in Mesquite, type in the sequence
into the text box and you will quickly be taken to it in
the taxon.
- Maintain
Clipboard Match (in the Find Sequence submenu of
the Edit menu): This is similar to Maintain Target Match,
except that it obtains the search string not from the text
area but from the clipboard. If the clipboard changes, the
function will automatically find the sequence again in the
matrix. This is useful if working with a program like Sequencher.
If you turn on Maintain Clipboard Match, then you can copy
stretches of a sequence within Sequencher, and Mesquite
will automatically highlight it, without your having to
return to Mesquite or give any other command to it. (Mesquite
is constantly monitoring the clipboard to see if it changes).
- There is an optional system for
assigning and maintaining ID strings to uniquely identify taxa
and characters. This can be turned on via the menu items Invent
Unique IDs for New Taxa and Invent Unique IDs
for New Characters in the Defaults submenu of the File menu. When enabled, newly created characters and taxa are
automatically assigned IDs which are maintained even if the
taxa or characters are rearranged through reordering, additions
or deletions. These unique IDs are little-used in Mesquite at
present, but eventually will permit users to reassociate taxa
and characters from different files even after names have been
changed (for instance, to re-read a tree file saved with different
taxon names). The IDs will also be used in database connectivity
and collaborative features. To assign IDs to already-created
taxa or characters, go to the List of Taxa or List of Characters
windows and choose Unique IDs from the Columns
menu. IDs can be generated using the drop down menu from the
column's header. The one disadvantage of assigning IDs is that
the IDs are stored in the Taxa block and Characters block of
the NEXUS file, which causes some programs to refuse to read
the file. (PAUP* by default will refuse to read these files,
but you can request that PAUP* ignore these extra commands by
the command "set errorstop = no;" which is available in its
Options Menu, Warnings and Errors.)
- There has been a shift in NEXUS
file format for annotations — the AN
command is now used. This, unfortunately, produces files that
are not readable by version 1.05. The reason for this change
is to minimize text in the file when dealing with a large matrix
with thousands of annotations.
- Pairwise comparisons
were previously forced to find all possible pairs. Users can
now impose a limit so that only the first n
pairs are found and considered. This is important if there are
too many pairs to efficiently consider.
- Support for the new Cartographer
package.
- Groups of characters and taxa can
now be assigned grayscale values instead of just colors.
- Rarefy Tree (a random tree modifier)
now uses taxon selection. If some of the taxa are selected,
than Rarefy Tree chooses which taxa to exclude randomly only
from among the selected taxa.
- There is a new option to export
files, Old-Fashioned NEXUS, for use by programs
that understand only a small subset of the NEXUS format such
as MrBayes.
- Trees can be drawn in "Eurogram"
style by using Square Tree and selecting the menu item Cut
Corners.
- The Developer's
documentation has been updated
Bug fixes
- ()
Fixed bug that caused labels for internal nodes (including Bayesian
support values) to be scrambled if the branch interchange or
reroot tool was used.
- ()
The PAUPConstraintTestParsimony template included with Batch
Architecture for doing the test of monophyly has one of the
searches incorrectly searching for the optimal trees with the
converse of the constraint rather than the constraint
itself; this has been fixed.
- ()
Fixed bug that caused tree statistics to be displayed incorrectly
in columns in the List of Trees window if trees were deleted.
- Fixed bug in dialog boxes with lists
from which to choose that made them unusable under Mac OS X
10.4.
- Fixed bug that prevented use of
high ASCII characters in NEXUS files (e.g. "é",
"ü", and so on).
- Fixed bug that greatly slowed file
reading under Java 1.5
- Fixed various issues to improve
performance under Mac OS X Java 1.4. (Some windows are still
misdrawn, but that is due to bugs in Mac OS X.)
- Fixed other minor bugs (including,
but not only: table copy/paste under Windows; use of arrow keys
in matrix under Windows; retention of full character information
in concatenated matrices; stepmatrix reading when character
matrix ambiguous; fill tool with gaps)
Version 1.05
New Features
- added a new Find feature (in the
Edit menu) that allows you to search for sequences within DNA
data matrices. The first example of a module doing this is "Matching
Sequence" which finds sequences matching a target
sequence the user enters. The search allows a certain number
of mismatches. Optionally, it can search for the reverse, complement
and reverse complement of the target sequence.
- added a new item in the Select menu
of Character Matrix Editor, "Select Lowercase Ends".
This selects the ends of a DNA sequence up to the first block
of N sites in a row with uppercase symbols. The user chooses
N. Assuming that lowercase letters are used for less certain
base calls, this can be used to select and then trim (by painting
with gaps) poorly-sequenced terminal regions.
- the Concatenate Matrices
command will now allow molecular sequence data to be concatenated
to the end of a matrix of standard categorical characters.
- in the Mac OS X download, added
an executable to run Mesquite under Java 1.4. This may be less
stable than the standard version running under Java 1.3.1. The
Java 1.4 version should be used only if you have Java 1.4.2
update 2 or later on Mac OS X. (The problems with early versions
of Java 1.4 appear to be restricted to Mac OS X.)
Bug Fixes
- ()
fixed bug in the Brownian motion simulations of continuous character
evolution. Brownian motion puts more change on long branches,
less change on short branches. The bug in the simulations caused
this contrast between long and short branches to be heightened
— long branches had even more change relative to short
branches than they should have. (Inadvertantly the effect of
branch length had been squared in the calculation.)
Version 1.04
New Features
- added Utilities submenu to Tree
menu of Tree Window. Currently there is only one utility, E-mail
Tree, which prepares a text image of the tree and puts
it in the body of an e-mail message requested using a web browser's
mailto: command.
Bug fixes
- ()
fixed bug in two tree simulators, Uniform speciation
(Yule) and Uniform Speciation with Sampling.
In versions 1.03 and before, these failed to randomize tip labels,
resulting in trees whose shapes matches the model's expectations,
but whose particular relationships did not. Thus, in previous
versions the taxa were added one at a time to a tree growing
through time. For example, if there are 6 taxa, then the divergence
between the 6th taxon and its sister species was always the
most recent divergence in the tree. While this would not affect
questions about general tree symmetry (e.g. "What is the
distribution of tree asymmetries under uniform speciation?")
or branch length distributions, it would affect questions about
similarity between an observed tree and those expected under
a null model of speciation.
- fixed bug that caused a crash when
characters were deleted from a matrix which had annotations,
images or colors attached to the cells but which didn't have
footnotes.
- fixed a few minor bugs.
Version 1.03
New Features
- added automatic file backup,
which retains copies of previously saved versions of files.
This feature was added to allow the user to return to a previous
version in case of errors or other problems. To enable this,
in the Log window or Projects & Files window select Automatic
NEXUS backup in the Defaults submenu of the File menu. Indicate the number of backups
to be saved. If you enter, for instance, 3, then when you save
a file named "myfile", the previous version of the
file (if any) will be retained and named myfileBKP1. It will
be in the same directory as myfile. The version before that
will be called myfileBKP2, the version before that myfileBKP3.
When you save the files again, the file ending in BKP3 will
be deleted, BKP2 will be named BKP3, BKP1 renamed BKP2, and
the newest backup will be called myfileBKP1.
- enabled grayscale shading
in Trace Character History for continuous-valued characters.
This can be turned on in the Trace menu by selecting the "Use
Grayscale If Continuous" menu item.
- Matrices can be compared by selecting
cells that differ. To request this, choose "Select
by Matrix Comparison" from the Select menu of the character
matrix editor. This is similar to the Utility "Compare
matrices", but instead of giving a textual summary of the
differences between two matrices, "Select by Matrix Comparison"
highlights those cells of the matrix that differ.
Bug fixes
- fixed a bug that caused NOTES blocks
to be written in duplicate form into all linked files (opened
via Link File...) if all files were saved. This bug was generally
innocent except if a linked file was subsequently unlinked from
the main data file. This would leave the linked file with a
relictual copy of the NOTES block. If the main data file was
then edited (e.g., footnotes changed, characters deleted or
moved), and then the secondary file was later linked again to
the main data file, its relictual (and now out of date) NOTES
block could override the current NOTES block in the main file.
This could cause footnotes in the main data file to revert to
former ones, or to be applied to inappropriate characters or
taxa. In addition to fixing this bug, we have added a warning
that is given if Mesquite detects that a NOTES block is being
overridden. If you suspect that your footnotes may have been
corrupted by this bug, please contact us for assistance.
- fixed a bug in Trees Directly from
File (now called "Use Trees from Separate File") that gave spurious warnings.
- fixed various other small bugs
Version 1.02
New Features
- multiple notes with images
can now be associated with taxa, characters and cells of character
matrices. Prior to 1.02 a single image could be associated with
each taxon, or a single footnote could be associated with each
taxon, character or cell of the matrix. Now, multiple notes
can be attached to each taxon, character, or cell of a matrix.
Each note can have a comment, a reference, an image, and an
author. Labels can be attached to the image. To use this,
select the notes tool ()
in the Character Matrix Editor. If you touch on a cell, a Annotations
window will appear with the notes for that cell, if any.
Buttons in this window can be used to delete and add notes and
images, and to make additional notes windows. Behavior of the
window can be modified using its Annotations menu. NOTE: the
single footnote system still exists and is independent of this
multiple notes system.
- MRP matrices can
be generated from a set of trees, for use by Matrix Representation
with Parsimony analyses to construct supertrees. To use this,
select MRP Matrices from trees as a source of matrices (e.g.,
under Characters menu, Make New Matrix submenu).
- a search facility
has been added to the Character Matrix editor and to the List
windows. The cell of the matrix found by the search is highlighted
and made visible. To use this, select Find String or
Find Footnote from the Edit menu. Find String searches through
the row names first (e.g., taxon names for the Character Matrix
Editor), then the column names (e.g., character names), then
the internal cells of the table (e.g., the character state codings).
Find Footnote searches the footnotes of the Character Matrix
and highlights cells containing the requested text in their
footnotes. You can also search for stretches of sequences or
state distributions matching the currently selected stretch
using Select Same Sequence and Select Same Distribution of the
Select menu of the Character Matrix Editor.
- More choices were added for coloring
character matrices. Both text and background of a cell
can now be colored according to properties of the cell. A paintbrush
tool (
 ) was
added to allow arbitrary coloring of cells of a character matrix. ) was
added to allow arbitrary coloring of cells of a character matrix.
- taxa and characters can
be moved by a menu item "Move Selected [taxa or
characters]To..." in the List of Taxa and List of Characters
windows. Prior to 1.02 taxa and characters could be moved by
clicking and dragging, but using this it was difficult to move
them long distances through a matrix. To use this,
select the taxa or characters to be moved in their respective
List window, and select Move Selected from the List menu.
- Taxon selecting in the tree window
has been modified, with a new taxon select tool (
 ). ).
- Utilities to modify taxon
names have been enhanced and added to the Character
Matrix Editor in the Taxon Names submenu of the Matrix menu.
- Added import/export of FASTA
files
Bug Fixes
- ()
fixed calculation of Shared Partitions, which counted too many
shared partitions between trees in some circumstances if the
trees had different terminal taxa included
- resolved conflict in interpetation
of tree descriptions that would result in Mesquite's interpreting
trees from MrBayes and other programs as having reticulations
whenever labels for internal nodes were duplicated. Mesquite
now interprets these labels as cosmetic only, and hence not
as indicating reticulations
- fixed bug involving the State Names
Editor, by which a character might mistakenly receive a name
duplicated from another character
- fixed bug in moving taxa or characters
by dragging past the end of the matrix
- fixed bug in recoding characters
by which state names and footnotes would not be adjusted
- fixed various other small bugs
Version 1.01
New Features
- added on-the-fly filtering
and transforming of trees. Thus, trees being supplied
from a file or simulation can be filtered according to various
criteria or can be transformed with the available tree or branch
length alterers. To use this, whenever a source of
trees is needed, choose first "Filter Trees from Other
Source" (if you want to select trees satisfying a criterion;
more details here) or "Transform
Trees from Other Source" (if you want to transform trees,
e.g. reroot, scale branch lengths, etc.; more details here),
then indicate what source of trees is to be filtered or transformed,
and what filter or transformation is to be used.
- added module to read trees
directly from a file one at a time; allows processing
of much larger tree files (>50,000 trees). To use this,
request "Use Trees from Separate File" as your source
of trees for charts and other calculations. You can also build
a tree block from a sample of trees from a large file by selecting
"Include partial contents" from the "Import File
With Trees" submenu of the Taxa&Trees menu.
- available tree and branch length
alterers can be applied to all trees in a tree block. To
use this, go to the Utilities submenu of the List menu
of the List of Trees window.
- added a new tree alterer: outgroup
rerooting of trees. To reroot trees using a selected
set of outgroups, you first must select the outgroup taxa, then,
choose the menu item "Root tree with selected taxa as outgroup",
for instance in the Alter/Transform Tree submenu of the Tree
menu of the tree window. More details are given on the page
on trees.
- added integrated BLAST search.
To use this, go to a character matrix editor window
showing a molecular matrix. Select one or more stretches of
sequence and choose BLAST Search from the Search submenu of
the Matrix menu. Mesquite will then send a request to your web
browser to perform a BLAST search at NCBI's BLAST server, one
request for each of the selected stretches of sequence. The
results will be available in your web browser.
- added -w (windowless)
and -b (backgroundable) flags for running long
Mesquite jobs in the background in Unix from the command line
- improved user interface of tables
(List Windows, Character Matrix Editor), including navigation
with arrow keys and more informative explanations and footnotes
- Added autotab
to the Character Matrix Editor. With this, the edited cell shifts
automatically to the right or downward when a state is entered.
To use this, select the Select and Type tool (with
the letter "K") and use its drop down menu to indicate
autotab right or autotab down. Autotab functions only when the
Select and Type tool is the active tool.
- selection of taxa, characters and
trees is now remembered when files are saved
- calculations of charts and Trace
Character Over Trees can now be stopped part way to show partial
results
Bug fixes
- fixed tree printing bug by which
node numbers and branch lengths always appeared
- fixed bug by which pressing the
Delete key while editing in a table would be misinterpreted
as a requesting to delete a character, taxon or other object
- fixed bugs in NEXUS file reading
and writing (concerning SYMBOLS list, blank character names,
tokenization with tab or square brackets, near-duplicate taxa
blocks in same file)
- fixed bugs in State Names Editor
and Character Matrix Editor concerning footnote display and
editing
- fixed bug with Mac OS X 10.3 by
which dialog boxes with lists always chose first item (this
was not a bug in Mesquite, but rather in Mac OS X, but we have
found a workaround)
- fixed bug with Windows by which
pie diagrams would be misdrawn as entirely black for traced
characters in the Balls and Sticks tree drawing mode (this was
not a bug in Mesquite, but rather in Windows/Java, but we have
found a workaround)
- several other smaller, less notable
bugs were also fixed
Release Dates
| 2.74 |
3 October 2010 |
| 2.73 |
24 July 2010 |
| 2.72 |
11 December 2009 |
| 2.71 |
7 September 2009 |
| 2.7 |
27 August 2009 |
| 2.6 |
24 January 2009 |
| 2.5 |
9 June 2008 |
| 2.01 |
7 December 2007 |
| 2.0 |
21 September 2007 |
| 1.12 |
23 September 2006 |
| 1.11 |
21 June 2006 |
| 1.1 |
18 May 2006 |
| 1.06 |
30 August 2005 |
| 1.05 |
24 September 2004 |
| 1.04 |
1 September 2004 |
| 1.03 |
1 July 2004 |
| 1.02 |
6 May 2004 |
| 1.01 |
14 January 2004 |
| 1.0 |
22 September 2003 |
|
|