|
|
|
Research
We use rational design and directed evolution to engineer novel protein molecules for use in biotechnology, research, and medicine.
Homology modeling, yeast surface display, directed evolution are used in our studies.
The current projects include the engineering and application of:
1. Monomeric streptavidin
2. Therapeutic proteins and antibodies
3. Antibody binding proteins
4. Others
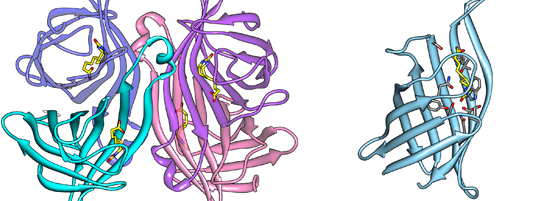
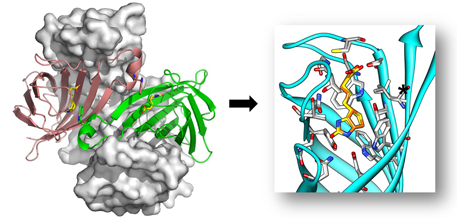
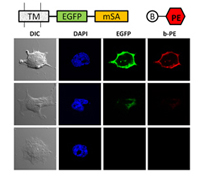
Engineered streptavidin monomer binds biotinylated ligands without crosslinking.
Addgene depositor page for monomeric streptavidin plasmids.
Purified protein is available at Sigma-Aldrich.
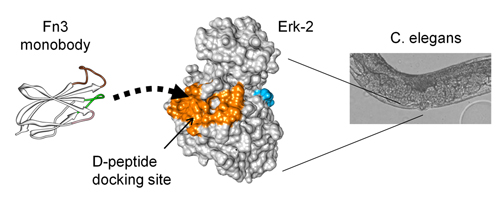
Engineered fibronectin derived monobodies (Fn3) bind the Erk-2 kinase at a protein-protein interaction site and inhibit its function. They are used to inhibit Erk-2 signaling in vitro, in mammalian cells, yeast, and C. elegans.
|