 |
|
|
|
|
Research Projects:: Catalysis of Proton and Hydride Transfer by Metal CationsHydride Transfer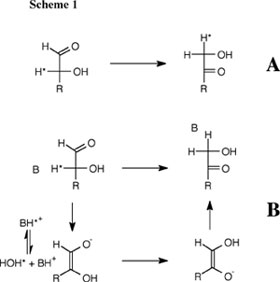
Aldose-ketose isomerization is a simple but mechanistically ambiguous reaction which involves the formal transfer of H2 from C-2 and O-2 to C-1 and O-1 of an α-hydroxy aldehyde to form the corresponding α-hydroxy ketone (Scheme 1). One hydrogen is transferred as a proton between the electronegative oxygens of the hydroxyl and carbonyl groups of the aldose. By contrast, transfer of the second hydrogen from C-2 to C-1 may occur directly as a hydride ion (Scheme 1A) or as a proton, in which case the bonding electron pair at C-2 moves through the carbon skeleton to C-1 (Scheme 1B). The question of the preferred pathway for this reaction in water has important implications in both Chemistry and Biology. We have shown that both both isomerization with hydride and with proton transfer can occur in aqueous conditions and that zinc dication stabilizes the transition state for both reactions, but provides a larger stabilization of the transition state for isomerization with hydride transfer. These data show that there is a clear imperative for metal ion catalysis of isomerization with hydride transfer, and provide a simple rationalization for the requirement of sugar isomerases for metal dications. See (a) Journal of the American Chemical Society, 118, 7432-7433 (1996) and (b) Journal of the American Chemical Society, 123, 794-802 (2001). Proton Transfer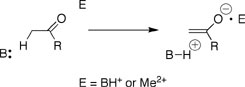
Proton transfer from carbon acids to Broensted bases is a simple chemical reaction that is a vital step of many complex biological processes. Proton transfer is strongly catalyzed by enzymes, but, as for many other simple reactions, there is no consensus about the origin of the enzymatic rate acceleration. Deprotonation of α-carbonyl carbon gives a delocalized "carbanion" where the negative charge lies mainly at the enolate oxygen that is derived from the neutral carbonyl oxygen at the substrate. This negative charge provides a target for stabilization by interaction with Broensted acids or metal ion electrophiles in catalyzed reactions. There have been several studies of metal ion catalysis of the deprotonation of relatively acidic carbon acids such as 1, which are activated for proton transfer by a second substituent (e.g. a phenyl group) and contain a basic site for chelation the metal ion. By comparison, there are only limited data for metal ion catalysis of the slower deprotonation of weakly acidic α-carbonyl carbon acids that are relevant models for enzyme-catalyzed deprotonation of keto sugars. The lack of model studies of metal ion catalysis of proton transfer from simple carbon acids has impeded discussions of the relative advantages of Broensted acid and metal ion catalysis by enzymes. 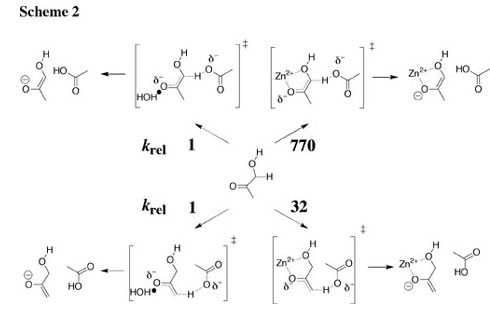
The deprotonation of the α-CH3 and α-CH2OD groups of hydroxyacetone and the α-CH3 groups of acetone in the presence of acetate buffer and zinc chloride in D2O at 25 C was followed by monitoring the incorporation of deuterium by 1H NMR spectroscopy, and the rate laws for catalysis of these reactions by acetate anion and zinc dication were evaluated. Relative to solvent water at a common standard state of 1 M, Zn2+ provides 6.3 and 4.4 kcal/mol stabilizations, respectively, of the transition states for deprotonation of the α-CH2OD and α-CH3 groups of hydroxyacetone by acetate anion (Scheme 2), and a smaller 3.3 kcal/mol stabilization of the transition state for deprotonation of the α-CH3 group of acetone. There is only a 1.4 kcal/mol smaller stabilization of the transition state for the acetate-ion-promoted deprotonation of acetone by the Broensted acid acetic acid than by Zn2+, which shows that, in the absence of a chelate effect, there is no large advantage to the use of a metal dication rather than a Broensted acid to stabilize the transition state for deprotonation of α-carbonyl carbon. See Journal of the American Chemical Society, 126, 5164-5173 (2004). |
| |||||||||
All material © John Richard 2003-2004 | Website Developed by Iserloh Design | ||||||||||