 Jian
Feng, Ph.D.
Jian
Feng, Ph.D.
SUNY Distinguished Professor
Department
of Physiology and Biophysics
School of Medicine &
Biomedical Sciences
State University of New York at
Buffalo
955 Main Street, Room 3148
Buffalo, NY 14203
Email: jianfeng@buffalo.edu
Phone: (716) 829-2345
Research:
My research is aimed at finding
the cause and a cure for Parkinson's disease.
Parkinson's disease (PD) is
defined by a characteristic set of locomotor symptoms (rest tremor, rigidity,
bradykinesia and postural instability) that are believed to be caused by the
selective loss of dopaminergic (DA) neurons in substantia nigra. The persistent
difficulties in using animals to model this human disease suggest that human
nigral dopaminergic neurons have certain vulnerabilities that are unique to our
species.
One of our unique features is
the large size of the human brain (1350 grams on average) relative to the body.
A single nigral dopaminergic neuron in a rat brain (2 grams) has a massive axon
arbor with a total length of 45 centimeters. Assuming that all mammalian
species share a similar brain wiring plan, we can estimate (using the cube root
of brain weight) that a single human nigral dopaminergic neuron may have an
axon with gigantic arborization that totals 4.6 meters.
Another unique feature of our
species is our strictly bipedal movement, which is affected by Parkinson's
disease, in contrast to the quadrupedal movement of almost all other mammalian
species. The much more unstable bipedal movement may require more dopamine,
which supports the neural computation necessary for movement.
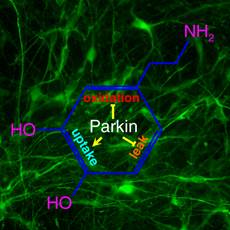 The landmark discovery of human induced pluripotent
stem cells (iPSC) made it possible to generate patient-specific human midbrain
dopaminergic neurons to study Parkinson's disease. A key problem for
dopaminergic neurons is the duality of dopamine as a signal required for neural
computation and a toxin as its oxidation produces free radicals. Our study
using iPSC-derived midbrain dopaminergic neurons from PD patients with parkin
mutations and normal subjects shows that parkin sustains this necessary duality
by maintaining the precision of the signal while suppressing the toxicity.
Mutations of parkin cause increased spontaneous release of dopamine and reduced
dopamine uptake, thereby disrupting the precision of dopaminergic transmission.
On the other hand, transcription of monoamine oxidase is greatly increased when
parkin is mutated. This markedly increases dopamine oxidation and oxidative
stress. These phenomena have not been seen in parkin knockout mice, suggesting
the usefulness of parkin-deficient iPSC-derived midbrain DA neurons as a
cellular model for Parkinson's disease. We are using iPS cells and induced DA
neurons to expand our studies to idiopathic Parkinson's disease. The
availability of human midbrain DA neurons should significantly speed up the
discovery of a cure for Parkinson's disease.
The landmark discovery of human induced pluripotent
stem cells (iPSC) made it possible to generate patient-specific human midbrain
dopaminergic neurons to study Parkinson's disease. A key problem for
dopaminergic neurons is the duality of dopamine as a signal required for neural
computation and a toxin as its oxidation produces free radicals. Our study
using iPSC-derived midbrain dopaminergic neurons from PD patients with parkin
mutations and normal subjects shows that parkin sustains this necessary duality
by maintaining the precision of the signal while suppressing the toxicity.
Mutations of parkin cause increased spontaneous release of dopamine and reduced
dopamine uptake, thereby disrupting the precision of dopaminergic transmission.
On the other hand, transcription of monoamine oxidase is greatly increased when
parkin is mutated. This markedly increases dopamine oxidation and oxidative
stress. These phenomena have not been seen in parkin knockout mice, suggesting
the usefulness of parkin-deficient iPSC-derived midbrain DA neurons as a
cellular model for Parkinson's disease. We are using iPS cells and induced DA
neurons to expand our studies to idiopathic Parkinson's disease. The
availability of human midbrain DA neurons should significantly speed up the
discovery of a cure for Parkinson's disease.
In our quest for making human
nigral DA neurons, we come to realize that these special cells are best made in
vivo, due to their extraordinary axon arborization. We have discovered a method
to convert human pluripotent stem cells (hPSCs) from the primed state to the
naive state. These naive hPSCs are cultured in essentially the same condition
used for maintaining mouse embryonic stem cells. When naive hPSCs are
transferred to mouse blastocysts, they generate up to 4% of mature human cells
of all three germ layers in mouse embryos at E17.5. A large amount of
enucleated human red blood cells are generated in mouse embryos, showing a
significant acceleration in the development of naive hPSCs in mouse embryos.
This technology enables the generation of human cells, tissues or even organs
in animals. By studying how human cells are made in chimeras, the long-term
goal is to generate human cells in an artificial system without the use of
animals.
Selected
Publications from my laboratory: (complete
bibliography)(Google
Scholar)
(44)
Z Jiang, K Saleem, E Fisher, L Li, Z Yan, J Feng (2026).
SGK1
inhibition restores excitability of cortical neurons derived from Alzheimer's
disease patients.
Neurobiology
of Disease 221:107345. [PDF]
(43)
E Fisher, Z Jiang, L Li, G Chhetri, K Saleem, Z Yan, J Feng (2026).
ASCL1
promotes nuclear shrinkage in transdifferentiation by suppressing NUP37.
Stem
Cell Reports 21:102823. [PDF]
(42) L Li, B Zhu, J Feng (2025).
Identifying
Key Regulators in Neuronal Transdifferentiation by Gene Regulatory Network
Analysis.
PNAS
Nexus 4:pgaf365 [PDF]
(41)
K Saleem, Z Xiao, B Zhu, Y Ren, Z Yan, J
Feng (2025).
Elevated
SGK1 increases Tau phosphorylation and microtubule instability in Alzheimer's
patient-derived cortical neurons.
Molecular
Psychiatry [PDF]
(40)
H Jiang, Z Xiao, K Saleem, P Zhong, L Li, G Chhetri, P Li, Z Jiang, Z Yan, J Feng (2025).
Generation
of human induced pluripotent stem cell-derived cortical neurons expressing the
six tau isoforms.
Journal
of Alzheimer's Disease 105:1341-1354 [PDF]
(39)
Zhu B, Fisher E, Li L, Zhong P, Yan Z, Feng
(2023)
PTBP2
attenuation facilitates fibroblast to neuron conversion by promoting
alternative splicing of neuronal genes.
Stem
Cell Reports 18:2268-2282 [PDF]
(38)
J Pu, L Lin, H Jiang, Z Hu, H Li, Z Yan, B Zhang, J Feng (2023)
Parkin
Maintains Robust Pacemaking in Human iPSC-derived A9 Dopaminergic Neurons.
Movement
Disorders 38:1273-1281 [PDF] [MDS
Podcast]
(37) H Li, H Jiang, H Li, L Li, Z Yan, J Feng (2022).
Generation
of human A9 dopaminergic pacemakers from induced pluripotent stem cells.
Molecular
Psychiatry 27:4407-4418. [PDF]
(36)
B Zhang and J Feng (2022).
Mouse
embryonic stem cells require multiple amino acids.
Experimental
Biology and Medicine DOI:
10.1177/15353702221096059. [PDF]
(35)
E Fisher and J Feng (2022).
RNA
Splicing Regulators Play Critical Roles in Neurogenesis.
Wiley
Interdisciplinary Reviews RNA
e1728. doi: 10.1002/wrna.1728 [PDF]
(34)
Y Ren, H Jiang, J Pu, L Li, J Wu, Y Yan, G Zhao, TJ Guttuso, B Zhang, J Feng
(2022).
Molecular
Features of Parkinson's Disease in Patient-Derived Midbrain Dopaminergic
Neurons.
Movement
Disorders 37:70-79.
[PDF]
(33)
B Zhang, H Li, Z Hu, H Jiang, AB Stablewski, BJ Marzullo, DA Yergeau, J Feng (2021).
Generation
of mouse-human chimeric embryos.
Nature
Protocols 16:3954-3980 [PDF]
(32)
B Li, H Jiang, H Li, B Zhang, M Slaughter, Z Yan, J Feng (2021).
Direct
conversion of adult human retinal pigmented epithelium cells to neurons with
photoreceptor properties.
Experimental
Biology and Medicine. 246:240-248
[PDF file]
(31)
J Feng (2021)
Modeling
the Pathophysiology of Parkinson's Disease in Patient-specific Neurons.
Experimental
Biology and Medicine. 246:298-304
[PDF file]
(30)
Z Hu, H Li, H Jiang, Y Ren, X Yu, J Qiu, AB Stablewski, B Zhang, MJ Buck, J Feng (2020).
Transient
Inhibition of mTOR in Human Pluripotent Stem Cells Enables Robust Formation of
Mouse-Human Chimeric Embryos.
Science
Advances 6: eaaz0298. [PDF file] [Press Coverage] [Altmetric]
(29)
H Li, Z Hu, H Jiang, J Pu, I Selli, J Qiu, B Zhang, J Feng (2020).
TET1
Deficiency Impairs Morphogen-free Differentiation of Human Embryonic Stem Cells
to Neuroectoderm.
Scientific
Reports 10:10343 [PDF file]
(28)
H Li, H Jiang, X Yin, JE Bard, B Zhang, J
Feng (2019).
Attenuation
of PRRX2 and HEY2 enables efficient conversion of adult human skin fibroblasts to
neurons.
Biochem
Biophys Res Commun. 516:765-769. [PDF file]
(27)
H Li, H Jiang, B Zhang, J Feng
(2018).
Modeling
Parkinson's Disease Using Patient-specific Induced Pluripotent Stem Cells.
Journal
of Parkinson's Disease 8:479-493. [PDF file]
(26)
P Zhong, Z Hu, H Jiang, Z Yan, J Feng
(2017).
Dopamine
Induces Oscillatory Activities in Human Midbrain Neurons with Parkin Mutations.
Cell
Reports 19:1033-1044. [PDF file]
(25)
Z Xu, X
Induced
dopaminergic neurons: A new promise for Parkinson's disease. Redox
Biology 11:606-612. [PDF file]
(24)
J Feng (2016).
Kinetic
Barriers in Transdifferentiation.
Cell
Cycle, 15:1019-1020 [PDF file]
(23)
Z Xu, H Jiang, P Zhong, Z Yan, S Chen, J
Feng (2016). Direct Conversion of Human Fibroblasts to Induced Serotonergic
Neurons. Molecular Psychiatry, 21:62-70. [PDF file]
(22)
H Jiang, Z Xu, P Zhong, Y Ren, G Liang, HA Schilling, Z Hu, Y Zhang, X Wang, S
Chen, Z Yan, J Feng (2015). Cell
Cycle and p53 Gate the Direct Conversion of Human Fibroblasts to Dopaminergic
Neurons. Nature Communications, 6: 10100. [PDF file]
(21)
Z Hu, J Pu, H Jiang, P Zhong, J Qiu, F Li, X Wang, B Zhang, Z Yan, J Feng (2015).
Generation
of Naivetropic Induced Pluripotent Stem Cells from Parkinson's Disease Patients
for High Efficiency Genetic Manipulation and Disease Modeling.
Stem
Cells and Development.
24:2591-2604. [PDF file]
(20) J Pu, D Frescas, B Zhang, J Feng (2015).
Utilization
of TALEN and CRISPR/Cas9 technologies for gene targeting and modification. Experimental
Biology and Medicine 240:1065-1070. [PDF file]
(19) Y Ren, H Jiang, Z Hu, K Fan, J Wang, S Janoschka,
X Wang, S Ge, J Feng (2015). Parkin
Mutations Reduce the Complexity of Neuronal Processes in iPSC-derived Human
Neurons. Stem Cells, 33:68-78. [PDF file]
(18) H Jiang, Y Ren, EY Yuen, P Zhong, M Ghaedi, Z Hu,
G Azabdaftari, K Nakaso, Z Yan, J Feng (2012).
Parkin Controls Dopamine Utilization in Human Midbrain
Dopaminergic Neurons Derived from Induced Pluripotent Stem Cells.
Nature Communications 3:668. [PDF file] [Press Coverage]
(17) Y. Ren, X. Liu, S.
Lesage, M. Cai, J. Pu, B. Zhang, A. Brice, J.
Feng (2011).
The
Movement Disorders PMID: 22095769. [PDF
file]
(16)
Y. Ren, H. Jiang, D. Ma, K. Nakaso, J.
Feng (2011).
Parkin
Degrades Estrogen Related Receptors to Limit the Expression of Monoamine
Oxidases.
Hum. Mol. Genet. 20:1074-1083. [PDF file]
(15) H. Jiang, D. Cheng, W.
Liu, J. Peng, J. Feng (2010).
Protein Kinase C Inhibits Autophagy and Phosphorylates
LC3.
Biochem Biophys Res Commun. 395:471-476. [PDF file]
(14)
Y. Ren, H. Jiang, F. Yang, K. Nakaso, and J.
Feng (2009).
Parkin
protects dopaminergic neurons against microtubule-depolymerizing toxins by
attenuating MAP kinase activation.
J.
Biol. Chem. 284:4009-4017. [PDF file]
(13)
Q. Jiang, Y. Ren, and J. Feng
(2008).
Direct
Binding with Histone Deacetylase 6 Mediates the Reversible Recruitment of
Parkin to the Centrosome.
J.
Neurosci. 28:12993-13002. [PDF file]
(12)
Y. Ren and J. Feng (2007).
Rotenone
Selectively Kills Serotonergic Neurons through a Microtubule-dependent
Mechanism.
J.
Neurochem. 103:303-311. [PDF file]
(11) J. Feng (2006).
Microtubule:
a Common Target for Parkin and Parkinson's Disease Toxins.
Neuroscientist. 12:469-476. [PDF file]
(10)
Q. Jiang, Z. Yan, and J. Feng
(2006).
Neurotrophic
factors stabilize microtubules and protect against rotenone toxicity on
dopaminergic neurons.
J.
Biol. Chem. 281:29391-29400. [PDF file]
(9) H.
Jiang, Q. Jiang, W. Liu and J. Feng (2006).
Parkin
Suppresses the Expression of Monoamine Oxidases.
J.
Biol. Chem. 281:8591-8599. [PDF file]
(8)
Q. Jiang, Z. Yan, and J. Feng
(2006).
Activation
of Group III Metabotropic Glutamate Receptors Attenuates Rotenone Toxicity on
Dopaminergic Neurons through a Microtubule-dependent Mechanism.
J.
Neurosci. 26:4318-4328. [PDF file]
(7)
Y. Ren, W. Liu, H. Jiang, Q. Jiang, and J.
Feng (2005).
Selective
Vulnerability of Dopaminergic Neurons to Microtubule Depolymerization.
J.
Biol. Chem. 280:34105-34112. [PDF file] [Media Coverage]
(6)
F. Yang, Q. Jiang, J. Zhao, Y. Ren, M.D. Sutton and J. Feng (2005).
Parkin
Stabilizes Microtubules through Strong Binding Mediated by Three Independent
Domains.
J.
Biol. Chem. 280:17154-17162.
[PDF file]
(5)
H. Jiang, Q. Jiang and J. Feng
(2004).
Parkin
Increases Dopamine Uptake by Enhancing the Cell Surface Expression of Dopamine
Transporter.
J.
Biol. Chem. 279:54380-54386. [PDF file]
(4)
H. Jiang, Y. Ren, J. Zhao and J. Feng
(2004).
Parkin
protects human dopaminergic neuroblastoma cells against dopamine-induced
apoptosis.
Hum. Mol. Genet. 13: 1745-1754. [PDF file]
(3) J. Feng (2003).
Genetic
factors in Parkinson's disease and potential therapeutic targets.
Curr.
Neuropharmacol. 1: 301-313. [PDF file]
(2)
J. Zhao, Y. Ren, Q. Jiang and J. Feng (2003).
Parkin
is recruited to the centrosome in response to inhibition of proteasomes.
J.
Cell Sci. 116: 4011-4019. [PDF file]
(1)
Y. Ren, J. Zhao and J. Feng (2003).
Parkin binds to a/β tubulin and
increases their ubiquitination and degradation.
J. Neurosci. 23: 3316-3324. [PDF file]
Biographical Information:
Education:
1993-1997:
Ph.D. Biochemistry (1997), research advisor: James N.
Ihle, Ph.D.
1986-1990: Nanjing University, Nanjing,
China.
B.Sc. Biochemistry (1990)
Academic Appointments:
2000-Present:
Professor (2010-), Associate Professor (2005-2010), Assistant Professor
(2000-2005)
Department of Physiology and Biophysics
1997-2000:
Postdoctoral Associate
Laboratory of Molecular and Cellular Neuroscience
The
Research advisor: Paul
Greengard, Ph.D.
Awards:
State
University of New York Distinguished Professor (04/25)
Stockton
Kimball Award and Lecture, the highest honor at the Jacobs School of Medicine
and Biomedical Sciences, University at Buffalo, the State University of New
York (05/24)
University at Buffalo Distinguished Professor (09/23)
State
University of New York Chancellor's Award for Excellence in Scholarship and
Creative Activities (05/21)
University at Buffalo
Exceptional Scholars - Sustained Achievement Award (10/17)
Top 100 Principle
Investigators, State University of New York at Buffalo (10/05).
Visionary Inventor Award,
Promising Inventor Award,
Top 100 Federal Grantee,
Young Investigator Achievement Award, SUNY-Buffalo
(5/02).
Theodore and Vada Stanley Foundation Research Award
(8/98-7/00).
Ralph R. Braund Young Investigator Award in Cancer
Research,
Alma and Hal Reagan Fellowship in Cancer Research,
Univ. of
Membership:
Parkinson's
Disease iPS Cell Line Consortium
International Society
for Stem Cell Research
Editorial Board:
07/12 - 06/27: Associate Editor (Stem Cell Biology
section),
Experimental
Biology and Medicine
Publications or media presentations offering
professional expertise:
(1) J. Feng. Entropy
illustrates the flexibility of Chinese. Nature 410: 1021 (2001)
http://www.nature.com/nature/journal/v410/n6832/pdf/4101021d0.pdf
(2) J. Feng.
Parkinson's Disease: Shootout at the Microtubule Corral?
The American Society for Cell
Biology Press Book 2004, page 8.
http://www.ascb.org/files/2004pressbook.pdf
(3) Interviewed about our work on novel
neuroprotective agents against Parkinson's disease,
on WBFO, an NPR station in
Buffalo, on Nov. 11, 2005.
http://www.publicbroadcasting.net/wbfo/news.newsmain?action=article&ARTICLE_ID=841528
(4) Interviewed about the potential risks of using
rotenone to control invasive fish,
on WKNO, an NPR station for
the Mid-South region, on Sept. 9, 2008. http://www.publicbroadcasting.net/wkno/news.newsmain?action=article&ARTICLE_ID=1360579
(5) Interviewed about the ban of federal funding of
embryonic stem cell research by Buffalo
Business First on September 3-9, 2010 issue.
http://buffalo.bizjournals.com/buffalo/stories/2010/09/06/story10.html
(6) J. Feng.
Embryonic stem cells: don't let litigation put research off limits. Nature 467: 271 (2010)
http://www.nature.com/nature/journal/v467/n7313/pdf/467271a.pdf
(7) Research Interview published in ''Parkinson's
Diseases World Drug Industry and Market 2016-2026''
(8) Interviewed by Australian Broadcasting Corporation
on ''Chimeras in Medicine'' (at 20'10'')
https://www.abc.net.au/radionational/programs/healthreport/25-oct-segment/13828204
(9) Celebrating the 175th Anniversary of JSMBS and UB: The Impact of Stem Cells
https://medicine.buffalo.edu/175/forward-thinking.html
(10) Interviewed by Drug Discovery News for the
article ''Chimera research opens new doors to understanding and treating
disease''
(11)
Interviewed by Nature about two
papers published in Cell on rat-mouse
chimeras.
''Rat
neurons repair mouse brains - and restore sense of smell''
https://www.nature.com/articles/d41586-024-01222-1
(12)
Interviewed by Men's Health on ''What Stem Cell Treatments Can - and Can't -
Do''
https://www.menshealth.com/health/a62894108/stem-cell-uses-treatments/
(13) Interviewed by Men's Health on ''Living Near a
Golf Course Nearly Doubles Your Risk of This Devastating Disease - Doctors
break down the link between where you live and Parkinson's.''
Selected media coverage of my research:
(1)
New Study Finds Mouse
Embryonic Stem Cells Have No Special Requirement for Threonine
(2)
Massive Pacemaker
Cells Produced in Parkinson's Breakthrough
(3)
Scientists made a
mouse embryo that's 4% human -- the highest level of human cells in an animal
yet.
https://www.cnn.com/2020/05/21/us/human-mouse-chimera-hybrid-scn-trnd/index.html
(4)
Millions of Human
Cells Have Been Grown Inside Mice Embryos.
https://www.newsweek.com/millions-human-cells-have-been-grown-inside-mice-embryos-1503629
(5)
UB Researchers
Reproduce Brain Oscillations that Characterize Parkinson's. https://www.aau.edu/research-scholarship/featured-research-topics/ub-researchers-reproduce-brain-oscillations
(6)
These serotonin
neurons were built in a dish.
https://www.futurity.org/serotonin-neurons-977552/
(7)
Parkinson's
disease researchers discover a way to reprogram the genome to produce dopamine
neurons
(8)
Genetic
Parkinson's disease brain cells made in lab.
https://www.bbc.com/news/health-16913997
(9)
Stem cell hope for
millions with Parkinson's disease.
https://www.mirror.co.uk/news/technology-science/science/parkinsons-disease-hope-after-stem-676768
Public Lecture:
Jian Feng. Finding a cure
for Parkinson's disease. Oct. 19, 2011. [video]
This is the prelecture talk
before the Michael J. Fox's Distinguished Speaker Lecture at UB.
Documentary:
Parkinson's Disease Research
at UB, introduction to Michael J. Fox's speech at UB.